Uniwersytet Medyczny
im. Karola Marcinkowskiego
w Poznaniu
Jakub Szrama
Wpływ wybranego schematu insulinoterapii na profil
glikemiczny i przebieg ciężkiej sepsy chorych leczonych w
oddziale intensywnej terapii
Praca doktorska napisana pod kierunkiem
Prof. dr hab. Leona Drobnika
OŚWIADCZENIE
Niniejszym oświadczam, iż jestem autorem pracy ……… pt.: ………. ………. ……… ………... ...
Praca ta została przeze mnie napisana samodzielnie (bez jakiegokolwiek udziału osób trzecich), przy wykorzystaniu wykazanej w pracy literatury przedmiotu i materiałów źródłowych, stanowi ona pracę oryginalną nie narusza praw autorskich oraz dóbr osobistych osób trzecich i jest wolna od jakichkolwiek zapożyczeń.
Oświadczam również, że wymieniona praca nie zawiera danych i informacji, które zostały uzyskane w sposób niedozwolony prawem oraz nie była dotychczas przedmiotem żadnej urzędowej procedury związanej z uzyskaniem tytułu……….., a złożona przeze mnie dyskietka/płytą CD zawiera elektroniczny zapis przedstawionej przeze mnie pracy.
Jednocześnie oświadczam, że nieodpłatnie udzielam Uniwersytetowi Medycznemu im. Karola Marcinkowskiego w Poznaniu licencji do korzystania z wyżej wymienionej pracy bez ograniczeń czasowych i terytorialnych w zakresie obrotu nośnikami, na których pracę utrwalono przez: wprowadzanie do obrotu, użyczenie lub najem egzemplarzy w postaci elektronicznej a nadto upoważniam Uniwersytet Medyczny im. Karola Marcinkowskiego
w Poznaniu do przechowywania i archiwizowania pracy w zakresie wprowadzania jej do pamięci komputera oraz do jej zwielokrotniania i udostępniania w formie elektronicznej oraz drukowanej.
Imię i nazwisko………...
Moją pracę doktorską dedykuję
ukochanej żonie w podziękowaniu
za wsparcie i natchnienie
SPIS TREŚCI
1 Wstęp...5 2 Sepsa...7 2.1 Definicje...7 2.2 Epidemiologia...8 2.3 Patofizjologia...9 2.4 Wytyczne leczenia...103 Metabolizm glukozy w organizmie...11
3.1 Metabolizm glukozy w warunkach fizjologii...11
3.1.1 Szlaki metaboliczne glukozy w ustroju...11
3.1.2 Mechanizmy kontrolujące przemianę glukozy...14
3.2 Metabolizm glukozy w warunkach stresu...15
3.2.1 Stress hyperglycemia...15
3.2.2 Niekorzystne następstwa hiperglikemii...17
4 Metabolizm glukozy w kontekście dysfunkcji wybranych narządów...19
4.1 Choroby wątroby...19
4.2 Choroby nerek...20
4.3 Niewydolność nadnerczy związana z krytyczną chorobą - Critical Illness Related Corticoid Insufficiency...22
4.4 Niewydolność trzustki...23
5 Badania własne...25
5.1 Hipotezy badawcze...25
5.2 Materiał i metoda badań...26
5.3 Wyniki...29 5.4 Dyskusja...75 5.5 Wnioski...85 6 Streszczenie...86 7 Abstract...88 8 Piśmiennictwo...90
1
Wstęp
Wstrząs septyczny jest jedną z najczęstszych przyczyn niewydolności wielonarządowej, która odpowiada za prawie 50 % zgonów chorych w oddziale intensywnej terapii [1]. Według raportu z 2002 roku sepsa stanowi 10 przyczynę zgonów mieszkańców Stanów Zjednoczonych [2]. Rozpowszechnianie wiedzy na temat sepsy, jej epidemiologii, manifestacji klinicznej oraz leczenia bierze swój początek w lipcu 2001, kiedy to opublikowano badanie epidemiologiczne pokazujące, że w Stanach Zjednoczonych liczba zgonów z powodu uogólnionej infekcji jest zbliżona do liczby zgonów z powodu zawału serca [3]. Doniesienia takie przyczyniły się do powstania międzynarodowej inicjatywy określanej Surviving Sepsis Campaign, założonej w 2002 roku, której celem było między innymi zwiększenie świadomości i wiedzy na temat sepsy, wpływ na opinię publiczną oraz zmniejszenie śmiertelności z powodu sepsy o 25 % w ciągu 5 lat. Owocem tej kampanii są wytyczne SSC, przedstawiające obszerne informacje na temat standardów postępowania terapeutycznego w sepsie [4]. Autorzy wśród takich podstawowych elementów terapii, jak optymalizacja stanu hemodynamicznego czy kontrola źródła zakażenia, zwracają uwagę na terapię wspomagającą, czyli między innymi wentylację mechaniczną, terapię naerkozastępczą czy kontrolę glikemii. I to właśnie ten ostatni element stał się w ostatniej dekadzie źródłem zaciętych dyskusji i polemik klinicznych i autorskich, a to wszystko za sprawą kilku badań przynoszących sprzeczne rezultaty i wprowadzających zamęt w umysłach klinicystów na co dzień walczących z sepsą przy łóżkach chorych. Sepsa podobnie jak większość krytycznych stanów chorobowych powoduje rozchwianie fukcjonowania całego ustroju, powodując ogólnoustrojową odpowiedź obronną organizmu. Rezultatem tej walki jest hiperglikemia, czyli poziomy glukozy we krwi powyżej 180 mg/dl, u chorych nie cierpiących na cukrzycę. Zjawisko to określono mianem "stress diabetes", czyli cukrzycy towarzyszącej reakcji stresowej organizmu na ostry stan chorobowy. Z początku uważano zjawisko to za odpowiedź obronną organizmu i przypisywano mu korzystne skutki dla organizmu walczącego z chorobą, twierdząc, że do walki niezbędne jest przecież paliwo, czyli glukoza [5]. Lata 90-te XX wieku przyniosły serię badań klinicznych zgodnie z duchem evidence based medicine, które udowodniły szkodliwe następstwa hiperglikemii u pacjentów z zawałem mięśnia sercowego i udarem mózgu [6,7,8]. Opierając się na takich doniesieniach postanowiono przenieść ich założenia na grunt oddziałów intensywnej terapii i krytycznie chorych. W 2001 roku opublikowano przełomowe badanie tzw. Leuven Study (nazwa pochodzi od miasta Leuven w Belgii), które na następne 10 lat z zmieniło podejście do hiperglikemii u krytycznie chorych. Autorzy udowodnili bowiem, iż utrzymywanie niskich wartości
glikemii 80 - 110 mg/dl u chorych w intensywnej terapii powoduje zmniejszenie śmiertelności [9]. Zachwyt nad tym odkryciem wiązał się z faktem, iż dotychczas tylko dla niewielu interwencji terapeutycznych u chorych w stanie krytycznym udało się udowodnić zmniejszenie śmiertelności. Udało się to przykładowo dla wentylacji oszczędzającej płuca w ARDS, czy tzw. early goal directed terapy w sepsie. Badacze z Leuven mieli swoich następców, ale doniesienia z kolejnych badań i metaanaliz przynosiły sprzeczne rezultaty co do korzyści z utrzymywanie niskich wartości glikemii u krytycznie chorych. Kres entuzjastycznym wynikom badania z Leuven przyniosło badanie NICE SUGAR, które jednoznacznie wykazało, że glikemia w przedziale 80 - 110 mg/dl wiąże się z większą śmiertelnością chorych w oddziałach intensywnej terapii oraz pociąga za sobą znaczne ryzyko ciężkich hipoglikemii [10]. Badanie to okazało się na tyle istotne, że autorzy wytycznych SSC postanowili zmienić dotychczasowe zalecenia dotyczące kontrolii glikemii w sepsie, wydając nowe zmodyfikowane zalecenie dotyczące tylko zagadnienia kontroli poziomu glukozy u chorych septycznych [11]. Rekomendacja autorów dotycząca kontynuowania badań nad optymalnym poziomem glikemii u chorych z sepsą oraz gorąca dyskusja w literaturze ostatnich 10 lat dotycząca tego aspektu stały się inspiracją do podjęcia przeze mnie zmagań z zagadnieniem glikemii u krytycznie chorych z ciężką sepsą.
2
Sepsa
2.1
Definicja
Obecność w organizmie człowieka źródła infekcji stanowi bodziec do ogólnoustrojowej reakcji obronnej organizmu. Pojęcia takie jak SIRS, sepsa, ciężka sepsa czy wstrząs septyczny charakteryzują progresję ciężkości stanu chorego cierpiącego z powodu uogólnionego zakażenia [12]. SIRS, rozwijając angielski skrót oznacza Systemic Inflammatory Response Syndrome, będącą ogólnoustrojową odpowiedzią zapalną, reakcją układu immunologicznego na bodziec naruszający homeostazę organizmu ludzkiego. Reakcja ta towarzyszy nie tylko sepsie, ale i innym stanom chorobowym, takim jak ostre zapalenie trzustki, oparzenie, uraz, rozległy zabieg operacyjny, które angażując układ odpornościowy powodują określoną odpowiedź ustroju [12]. Termin SIRS został wprowadzony w roku 1992 w związku z trudnościami dotyczącymi definiowania sepsy, którą pierwotnie określano jako odpowiedź organizmu na znane źródło infekcji z obecnością bakterii we krwi [13]. Problemy z definiowaniem sepsy wynikały z faktu, że często mimo objawów klinicznych charakteryzujących reakcję obronną ustroju, nie udawało się potwierdzić obecności bakterii we krwi chorego. Zgodnie z postanowieniami konferencji uzgodnieniowej American College of Chest Physicians i Society of Critical Care Medicine z 1992 SIRS rozpoznajemy, gdy spełnione są następujące kryteria :
• temperatura ciała > 38,5 ° C lub < 36 ° C
• czynność serca > 90/min
• częstość oddechów > 20/min lub PaCO2 < 32 mmHg (prężność dwutlenku węgla we krwi tętniczej)
• liczba leukocytów > 12 x 109/L lub < 4 x 109/L lub > 10% niedojrzałych neutrofilii w rozmazie krwi obwodowej [14]
Na tej samej konferencji wprowadzono definicje sepsy, ciężkiej sepsy, wstrząsu septycznego. Sepsę rozpoznawano, gdy spełnione były dwa lub więcej kryteria SIRS oraz gdy znaleziono lub podejrzewano źródło infekcji. Definicja ta na konferencji International Sepsis Definitions Conference w 2001 roku została rozszerzona i uzupełniona o wybrane parametry kliniczne i laboratoryjne takie jak zaburzenia świadomości, istotne obrzęki lub dodatni bilans płynowy > 20ml/kg w ciągu 24 godzin, hiperglikemia > 110mg/dl u pacjenta nie chorującego wcześniej na cukrzycę, stężenie CRP > 2 SD wartości prawidłowych, stężenie prokalcytoniny > 2 SD wartości
prawidłowych [15]. Ciężką sepsę definiuje się jako sepsę, której towarzyszy dysfunkcja narządowa charakteryzowana hipoperfuzją narządową (podwyższony poziom mleczanów, oliguria, zaburzenia krążenia obwodowego, zaburzenia stanu świadomości), zaburzeniami ze strony układu krzepnięcia (trombocytopenia, rozsiane wykrzepianie wewnątrznaczyniowe), układu oddechowego (ARDS), nerek (ostra niewydolność nerek), układu pokarmowego z dysfunkcją wątroby. Wstrząs septyczny rozpoznawano, gdy hipotensja (skurczowe ciśnienie tętnicze < 90 mmHg lub spadek o > 40 mmHg w stosunku do wartości wyjściowej) indukowana sepsą utrzymywała się pomimo właściowej płynoterapii [16].
2.2
Epidemiologia
Pomimo spektakularnych postępów w diagnostyce i terapii sepsy pozostaje ona wiodącą przyczyną zgonów chorych w oddziałach intensywnej terapii (OIT). SIRS i sepsa dotycza około 750 tysięcy pacjentów rocznie w Stanach Zjednoczonych z wzrastającym odesetkiem zachorowań, rzędu 1,5% rocznie [17]. Śmiertelność w sepsie waha się od 30 - 70 %, przy kosztach dla budżetu służby zdrowia w Stanach Zjednoczonych około 16,7 biliona dolarów rocznie [18]. Badanie z 2000 roku obrazujące epidemiologię sepsy w Stanach Zjednoczonych w latach 1979 - 2000 pokazuje wzrost śmiertelności z powodu sepsy. Autorzy wyciągnęli wniosek, iż ciężka sepsa to częsta, kosztowna i związana z dużą śmiertelnością jednostka chorobowa, z roczną liczbą zgonów porównywalną z liczbą zgonów z powodu zawału serca [19]. W Wielkiej Brytanii 27,1 % chorych dorosłych przyjmowanych do oddziału intensywnej terapii rozwija sepsę w ciągu pierwszych 24 godzin pobytu [20]. Zgodnie z doniesieniami z 2003 roku zmienia się natura drobnoustrojów odpowiedzialnych za wywoływanie ciężkiej sepsy. Podczas gdy za większość zakażeń wewnątrzszpitalnych odpowiadały bakterie Gram ujemne, coraz częściej spotyka się infekcje bakteriami Gram dodatnimi (30 - 50 % przypadków) oraz szczepami wielolekoopornymi lub grzybami (25%) [19,21]. W okresie między grudniem 2004 a czerwcem 2005 roku przeprowadzono badanie mające na celu określić częstość sepsy w oddziałach intensywnej terapii w Polsce. Autorzy tego badania stwierdzili, iż około 35 % chorych oddziałów intensywnej terapii jest leczonych z powodu różnych form sepsy. Przewidywaną częstość zachorowania na ciężką sepsę określono jako 34 na 100 tysięcy osób. Z kolei wystąpienie dowolnej formy sepsy szacuje się jako 91 przypadków na 100 tysięcy osób. Uwzględniając fakt, że badanie objęło tylko chorych oddziałów intensywnej terapii rzeczywista częstość sepsy wydaje się być 2 - 4 krotnie większa [22].
2.3
Patofizjologia
Patofizjologiczna odpowiedź organizmu w sepsie opiera się na nadmiernej reakcji układu odpornościowego na bodziec wywołujący sepsę [23]. Kluczową rolę odgrywa zachowanie równowagi układu immunologicznego między jego funkcją obronną, a jego nadmierną reakcją prowadzącą do uszkodzenia własnych tkanek, która to równowaga jest zaburzona w SIRS i w sepsie [12]. W początkowym etapie sepsy dochodzi do aktywacji układu immunologicznego, zarówno jego mechanizmów komórkowych, jak i humoralnych [12]. Decydującą rolę w rozpoznaniu zakażenia i aktywacji kaskady układu odpornościowego odgrywa odporność wrodzona. Odpowiedź wrodzona w przypadku bakterii Gram ujemnych aktywowana jest obecnością LPS, czyli lipopolisachardu zwanego też endotoksyną, składnika ściany komórkowej tychże bakterii. LPS wiąże się ze specyficznym białkiem osoczowym LBP (LPS binding protein), po czym kompleks ten aktywuje receptor CD 14 na powierzchni makrofagów i komórek śródbłonka [24]. Z kolei odpowiedź na infekcję bakteriami Gram dodatnimi jest wtórna do produkcji swoistej egzotoksyny lub częściej jest reakcją na fragment błony komórkowej tych bakterii [24]. Odpowiedź układu immunologicznego wiąże się z aktywacją komórek śródbłonka, neutrofilów, makrofagów i limfocytów, które produkują mediatory prozapalne, takie jak czynnik martwicy nowotworów TNF α, interleukinę 6 i 8. Wzrasta stężenie białek ostrej fazy (CRP - białko C reaktywne), dochodzi do aktywacji układu dopełniacza z produkcją jego składowych C3a i C5a. Wzrostowi stężenia cytokin prozapalnych towarzyszy wzrost także mediatorów przeciwzapalnych. Komórki fagocytarne (neutrofile i makrofagi) w odpowiedzi na cytokiny uwalniają enzymy ze swoich ziarnistości i wytwarzają aktywne formy tlenu, które odpowiadają za śmierć bakterii. Reaktywne formy tlenu obok tlenku azotu (NO) odpowiadają za uszkodzenie tkanek, zwiększoną przepuszczalność naczyń i w konsekwencji niewydolność narządów [12]. NO produkowany jest przez dwie formy enzymu syntazy (NOS - NO synthase): konstytutywną cNOS (obecną w neuronach i w śródbłonku) i indukowaną iNOS (obecną między innymi w komórkach mięśni gładkich, makrofagach). Ekspresja iNOS stymulowana jest przez rekację zapalną, a enzym ten produkuję większe ilości NO niż forma konstytutywna cNOS. Nadmierna produkcja NO powoduje zaburzenie funkcjonowania łańcucha oddechowego w mitochondriach powodując powstawanie wolnych rodników tlenowych, które z kolei niszsząc komlpeks I i III łańcucha oddechowego prowadzą do apoptozy komórek. Dysfunkcja mitochondriów została potwierdzona w wielu tkankach podczas sepsy, między innymi w monocytach, komórkach błony śluzowej jelit czy wątrobie [24]. Aby ułatwić migrację komórek fagocytarnych do miejsca infekcji dochodzi do zwiększonej ekspresji molekuł adhezyjnych na
powierzchni komórek śródbłonka (ICAM-1, E selektyny) i neutrofilach (CD11b/CD18) [25]. Reakcjom tym towarzyszy także aktywacja układu krzepnięcia i fibrynolizy, których dysfunkcja może doprowadzić do rozwoju rozsianego wykrzepiania śródnaczyniowego - DIC (disseminated intravascular coagulation). Ten wczesny etap sepsy określany jest jako etap hiperdynamiczny i klinicznie charakteryzuje go tachykardia, wzrost rzutu serca i wzrost oporu naczyń obwodowych [12]. W późniejszym etapie sepsy produkowane są mediatory przeciwzapalne, między innymi IL -10 i 13, których rola polega na antagonizowaniu działania cytokin prozapalnych wytworzonych w wczesnej fazie sepsy. Interleukiny te powodują supresję czynnika NF-κβ, która skutkuje zmniejszeniem aktywacji określonych genów odpowiedzialnych za syntezę mediatorów prozapalnych [26]. Produkcja mediatorów przeciwzapalnych i ich działanie nosi miano CARS - compensatory anti-inflammatory response syndrome, w analogii do SIRS [27]. Progresja zakażenia i sepsy prowadzi do zmniejszonej aktywności fagocytarnej neutrofilów i makrofagów, do zmniejszonej produkcji rodników tlenu, mniejszej zdolności bakteriobójczej, zmniejszonej chemotaksji i produkcji cytokin. Mechanizmy prowadzące do tej immunosupresji nie są jeszcze w pełni poznane. Przestawienie się z aktywności prozapalnej na przeciwzapalną, rozwój stanu "anergii immunologicznej" (utrata zdolności układu odpornościowego do odpowiedzi zapalnej), apoptoza komórek układu odpornościowego oraz immunosupresyjny efekt limfocytów prowadzą do supresji całego układu immunologicznego pacjentów z sepsą [28]. Ten "paraliż" układu odpornościowego i hyporeaktywna odpowiedź obronna organizmu występujące w późnym etapie sepsy charakteryzują się klinicznie zmniejszonym rzutem serca, bradykardią i zmniejszonym oporem obwodowym naczyń krwionośnych [12]. Konsekwencją reakcji układu immunologicznego, zaburzeń układu krzepnięcia, upośledzenia funkcji komórek sródbłonka, zaburzeń metabolizmu i oddychania komórkowego jest rozwój niewydolności wielonarządowej [24,29].
2.4
Wytyczne leczenia
Celem powstania kampanii Survivng Sepsis Campaign było między innymi wprowadzenie ujednoliconych standardów terapeutycznych w sepsie. W wytycznych tych wyróżnione zostały dwie grupy postępownia terapeutycznego I - Managment of Severe Sepsis oraz II - Supportive Therapy of Severe Sepsis [4]. W pierwszej grupie zaleceń rekomenduje się tzw. goal directed therapy, czyli postępowanie terapeutyczne służące osiągnięciu określonych celów, wartości parametrów takich jak ośrodkowe ciśnienie żylne, średnie ciśnienie tętnicze, saturacja mieszanej krwi żylnej, diureza godzinowa. Podstawowe postępowanie w sepsie obejmuje resuscytację
początkową, wczesne rozpoznanie, identyfikację źródła infekcji, antybiotykoterapię empiryczną i celowaną, płynoterapię, stosowanie amin katecholowych, steroidów i rekombinowanego białka C (w świetle doniesień badania PROWESS SHOCK obecnie nie zaleca się stosowania rekombinowanego białka C). Kontrola glikemii obok odpowiedniej strategii wentylacji mechanicznej, terapii nerkozastępczej, przetoczeń preparatów krwiopochodnych, sedacji i analgezji, wymieniana jest w drugiej grupie zaleceń - terapii wspomagającej - Supportive Therapy. W odniesieniu do kontroli glikemii w 2008 roku zalecano stosowanie dożylnej insulinoterapii celem kontroli hiperglikemii zgodnie ze sprawdzonymi, zatwierdzonymi protokołami insulinoterapii oraz zalecono utrzymywanie glikemii poniżej 150 mg/dl. W czerwcu 2009 roku w związku z doniesieniami badania NICE SUGAR opublikowano nowe rekomendacje odnośnie kontroli glikemii w sepsie. Taka precedensowa zmiana mimo licznych badań klinicznych nad sepsą dotyczyła jedynie rekombinowanego białka C (badanie PROWESS SHOCK) oraz zagadnienia glikemii, co obrazuje jak istotne okazały się wyniki badania NICE - SUGAR, a także pokazuje, iż mimo wielu publikacji na temat glikemii w sepsie, kwestia ta pozostaje wciąż niezwykle problematyczna. Zgodnie z tymi zmodyfikowanymi rekomendacjami nie jest zalecana już ścisła kontrola glikemii i utrzymywanie glikemii w przedziale 80 - 110 mg/dl, gdyż wiadomo, że nie zmniejsza ona śmiertelności, a wiąże się z częstszymi epizodami hipoglikemii. Zaleca się włączanie terapii insuliną dożylną, gdy glikemia przekracza 180 mg/dl oraz utrzymywanie jej na poziomie około 150 mg/dl. Autorzy na wstępie swych doniesień podkreślają, że ilość informacji z randomizowanych kontrolowanych badań klinicznych jest niewystarczająca, aby zalecić konkretne docelowe wartości glikemii u pacjentów z sepsą [30,31].
3
Metabolizm glukozy w organizmie
3.1
Metabolizm glukozy w warunkach fizjologii
3.1.1. Szlaki metaboliczne glukozy w ustroju
Glukoza jest najważniejszym węglowodanem, ponieważ większość węglowodanów zawartych w pokarmach wchłania się do krwioobiegu jako glukoza lub jest przekształcana w nią w wątrobie, a w organizmie z glukozy mogą powstać wszystkie inne cukry. Glukoza stanowi źródło energii w tkankach, jest skłanikiem proteoglikanów tworzących istotę podstawową tkankek oraz glikoprotein obecnych w błonach komórkowych. Glukoza metabolizowana jest we wszystkich komórkach ustroju w procesie glikolizy do pirogronianu i mleczanu. Wyjątkowa rola glukozy jako substratu polega na tym, że może być metabolizowana także w nieobecności tlenu; wówczas produktem końcowym glikolizy jest mleczan. Przemiana glukozy w procesie glikolizy prowadzi do powstania acetylo-CoA, który wchodzi w cykl kwasu cytrynowego, ulegając całkowitemu utlenieniu i uwalniając znaczne ilości energii wykorzystywanej do syntezy ATP w procesie fosforylacji oksydacyjnej. Inne szlaki przemiany glukozy to synteza glikogenu - zapasowego polimeru glukozy, zwłaszcza w mięśniach szkieletowych; szlak pentozofosforanowy, który jest dostawcą równoważników redukujących do biosyntezy kwasów tłuszczowych (brak jednego z enzymów szlaku pentozofosforanowego prowadzi do niedokrwistości hemolitycznej) oraz jest źródłem rybozy do syntezy nukleotydów i kwasów nukleinowych. Produkty pośrednie przemian glukozy uczestniczą w tworzeniu acylogliceroli, aminokwasów, kwasów tłuszczowych i cholestorolu, będącego prekursorem wszystkich steroidów syntetyzowanych w organizmie. Z kolei w procesie glukoneogenezy wytwarzana jest glukoza z prekursorów niecukrowych, między innymi z mleczanu, aminokwasów i glicerolu.
Glukoza powstająca w wyniku trawienia węglowodanów jest wchłaniana z jelit do żyły wrotnej, którą kierowana jest do wątroby. Wątroba wychwytuje nadmiar glukozy i przekształca ją w glikogen (glikogenogeneza) lub w tłuszcz (lipogeneza). Między posiłkami, w celu uzupełnienia stężenia glukozy we krwi, wątroba uwalnia ją ze zmagazynowanego glikogenu (glikogenoliza) lub, wraz z nerką, przekształca metabolity niecukrowe, takie jak mleczan (cykl Cori - cykl kwau
mlekowego), glicerol i aminokwasy (cykl glukozowo-alaninowy), w glukozę (glukoneogeneza). Glukoneogeneza może zachodzić tylko w wątrobie i w nerce z uwagi na obecność w tych tkankach specyficznego enzymu - glukozo 6 fosfatazy, który przekształcając glukozo 6 fosforan w glukozę, umożliwia uwolnienie glukozy do krążenia z tych tkanek. Utrzymanie właściwego stężenia glukozy we krwi jest konieczne ze względu na tkanki takie jak mózg i erytrocyty, dla których jest ona obligatoryjnym źródłem energii [32,33].
Przenikanie glukozy do większości komórek odbywa się drogą dyfuzji ułatwionej, poprzez wiązanie się z błonowym białkiem nośnikowym, które przenosi cząsteczkę glukozy zgodnie z gradientem stężeń. Zidentyfikowano 5 rodzajów transporterów przezbłonowych glukozy - GLUT, z któych najważniejsze znaczenie mają GLUT 1, 2 i 4 [34]. GLUT 1 występuje w wielu tkankach i jest odpowiedzialny za przenikanie glukozy do komórek w warunkach podstawowej przemiany materii. GLUT 2 z kolei odpowiada za swobodne, dwukierunkowe przenikanie glukozy przez komórki wątroby oraz za zależną od glukozy sekrecję insuliny [35]. Glukoza po wniknięciu do komórek β trzustki wchodzi w szlak glikolityczny i wytwarzane są duże ilości ATP, który blokuje ATP-wrażliwe kanały potasowe, powodując depolaryzację błony komórek β, co poprzez wzrost stężenia jonów Ca 2+ napływających przez zależne od napięcia kanały wapniowe, powoduje egzocytozę insuliny [32]. Z kolei izoforma GLUT 4 obecna jest w tkance mięśniowej, tłuszczowej i w sercu, w których to tkankach pobieranie glukozy zależne jest od insuliny. Insulina pobudza migrację zlokalizowanego w cytoplazmie komórki transportera GLUT 4 do błony komórkowej [34]. W warunkach podstawowej przemiany materii 80 % całkowitego poboru glukozy przez tkanki odbywa się drogą transportu niezależnego od insuliny (z ang. NIMGU non insulin mediated glucose uptake), głównie przez komórki ośrodkowego układu nerwowego [36]. 20 % poboru glukozy przez tkanki odbywa się w tkance mięśniowej, z czego połowa jest to transport NIMGU, a połowa transport zależny od insuliny (z ang. IMGU - insulin mediated glucose uptake). Insulina nie ma bezpośredniego wpływu na wnikanie glukozy do komórki wątrobowej; wieksze znaczenie w tym aspekcie odgrywa stężenie glukozy w żyle wrotnej oraz aktywność enzymów wątrobowych (np. glukokinazy) oraz stężenie produktów pośrednich metabolizmu glukozy (glukozo - 6 fosforan) [37,38]; po dłuższym czasie jednak w wyniku działania na syntezę enzymów kontrolujących glikolizę, glikogenogenezę i glukoneogenezę wzmaga się pobieranie glukozy przez wątrobę [32].
3.1.2. Mechanizmy kontrolujące przemianę glukozy
Kontrola stężenia glukozy we krwi odbywa się przez interakcję mechanizmów hormonalnych, nerwowych oraz wątrobowych. Stężenie osoczowe glukozy jest ściśle regulowane, mimo znacznych wahań w dostarczaniu i zużywaniu glukozy przez organizm. Insulina obniża poziom glukozy we krwi poprzez nasilenie pobierania glukozy przez tkanki, przez wzmożenie syntezy glikogenu oraz zahamowanie glukoneogenezy. Z kolei glukagon, katecholaminy, kortyzol i hormon wzrostu zwiększają stężenie glukozy we krwi przez stymulację glikogenolizy oraz glukoneogenezy i hamowanie obwodowego zależnego od insuliny poboru glukozy [5]. Regulacja sekrecji insuliny różni się od regulacji sekrecji innych hormonów. W warunkach spoczynkowych stężenie insuliny w osoczu zmienia się szybko w odpowiedzi na zmiany poziomu glikemii. Z kolei hormony antagonistyczne pobudzane są przez hipoglikemię lub inne bodźce takie jak oparzenia, urazy lub sepsę [5]. Glukagon produkowany przez komórki α trzustki w odpowiedzi na hipoglikemię, powoduje wzmożoną glikogenolizę i glukoneogenezę. Hormon wzrostu z kolei zmniejsza pobieranie glukozy przez mięśnie, a długotrwałe podawanie hormonu wzrostu prowadzi do cukrzycy. Glikokortykosteroidy nasilają glukoneogenezę w wyniku zwiększonego katabolizmu białek, zwiększonego pobierania aminokwasów przez wątrobę, wzrostu aktywności enzymów glukoneogenezy; hamują one także zużywanie glukozy w tkankach pozawątrobowych. Adrenalina również wywiera działanie hiperglikemizujące poprzez wzmożenie glikogenolizy w wątrobie i mięśniach oraz przez nasilenie glukoneogenezy [32].
W ośrodkowym i obwodowym układzie nerwowym istnieją receptory monitorujące poziom glikemii. W odpowiedzi na hiperglikemię zwiększa się aktywność neuronów glukoreceptorowych jądra brzusznoprzyśrodkowego podwzgórza, co w konsekwencji powoduje zahamowania wpływu układu współczulnego na trzustkę i prowadzi do sekrecji insuliny. Z kolei hipoglikemia zwiększa wyładowania w jądrze pasma samotnego i glukoreceptorach bocznej części podwzgórza, co zwiększa aktywność układu współczulnego, prowadząc do podwyższenia poziomu glikemii przez pobudzenie glikogenolizy i hamowanie sekrecji insuliny. W układzie nerwowym obwodowym receptory dla glukozy w żyle wrotnej, wątrobie i w jelicie cienkim zmniejszają swoje wyładowanie w odpowiedzi na hiperglikemię. Ten sygnał jest przewodzony przez nerw błędny do podwzgórza i jądra pasma samotnego w rdzeniu przedłużonym, wskutek czego hamowana jest aktywność układu współczulnego, a pobudzany jest układ przywspółczulny co zwiększa wydzielanie insuliny i hamuje sekrecję katecholamin [39]. Sygnał o zwiększonym poziomie glikemii w żyle wrotnej dociera także do ośrodkowego układu nerwowego, w wyniku czego stymulowane jest wątrobowe pobieranie glukozy.
W kontroli poziomu glikemii odgrywa rolę proces autoregulacji wątrobowej, który prowadzi do zmniejszenia wątrobowej produkcji glukozy w odpowiedzi na wzrost glikemii osoczowej. Wątrobowa produkcja glukozy regulowana jest poprzez tworzenie glukozo - 6 fosforanu powstającego w procesie glikogenolizy, glukoneogenezy i glikolizy oraz jego defosforylację do glukozy. Hiperglikemia prowadzi do spadku wątrobowej produkcji glukozy o 60 %, zmniejszenia glikogenolizy wskutek wzrostu wewnątrzwątrobowego poziomu glukozo 6 fosforanu powstającego w wyniku działania glukokinazy na glukozę osoczową wnikającą do hepatocytów [40]. Autoregulacja wątrobowa zapobiega też nadmiernemu uwalnianiu glukozy do krążenia w sytuacji wzrostu stężenia prekursorów glukoneogenezy. Infuzja mleczanu lub glicerolu powoduje stymulację glukoneogenezy, ale nie zwiększa endogennej produkcji glukozy [41]. Wydaje się, że kluczową rolę w autoregulacji wątrobowej odgrywają wątrobowe zapasy glikogenu [42].
3.2
Metabolizm glukozy w warunkach stresu
3.2.1. Stress hyperglycemia
Związek między urazem a wzrostem poziomu glikemii w osoczu zauważył już w XIX wieku fizjolog francuski Claude Bernard. Zależność ta ma w literaturze różne określenia "stress diabetes", "traumatic diabetes", "diabetes of injury" oraz używane obecnie "stress hyperglycemia". U pacjentów, którzy wcześniej nie chorowali na cukrzycę, a którzy doznali zawału mięśnia sercowego stress hyperglycemia występuje u 3 do 71 % chorych [8]. Wzrost poziomu glukozy w osoczu obserwuje się także u około 50 % chorych z sepsą, którzy wcześniej nie chorowali na cukrzycę [43]. Odpowiedź organizmu na stres, ciężką chorobę czy uraz obejmuje aktywację ośrodkowego układu nerwowego, pobudzenie wydzielania hormonów stresu oraz mobilizację układu cytokin. Aktywacja ośrodkowego układu nerwowego prowadzi do pobudzenia osi podwzgórze- przysadka - nadnercza, układu współczulnego oraz rdzenia nadnerczy [44]. Kluczową rolę odgrywa tu podwzgórzowy hormon uwalniający kortykotropinę (CRH) oraz noradrenergiczne neurony miejsca sinawego, przysadki i pnia mózgu [44]. Pobudzenie osi podwzgórze-przysadka - nadnercza prowadzi do wzrostu produkcji kortyzolu przez korę nadnerczy. W regulacji tej osi znaczenie mają także cytokiny prozapalne (m.in TNF α, IL-1, IL-2, IL-6, INF α) uwalniane w sytuacji stresu, które pobudzają produkcję CRH, ACTH oraz kortyzolu bezpośrednio w nadnerczach [44,45,46]. Kaskada reakcji wywołana sytuacją stresową lub urazem prowadzi do wzrostu poziomu glukozy w osoczu.
Początkowo wskutek działania katecholamin dochodzi do pobudzenia glikogenolizy wątrobowej, ze szczytem tej reakcji w ciągu trzech godzin od urazu [47]. W tym początkowym etapie nie odgrywa istotnej roli glukagon, gdyż jego stężenia osoczowe bezpośrednio po urazie pozostają w normie [48,49]. Następnie dochodzi do wzmożonej produkcji wątrobowej glukozy w procesie glukoneogenezy. Tutaj większe znaczenie niż katecholaminom, przypisuje się glukagonowi [50] oraz kortyzolowi [51,52]. Stężenie osoczowe kortyzolu jest umiarkowanie podwyższone w następstwie urazu i wydaje się, że rolą kortyzolu jest potencjalizacja wątrobowego działania glukagonu i adrenaliny, stymulującego produkcję glukozy [5]. Podczas odpowiedzi stresowej na uraz dochodzi także do wzrostu poziomu hormonu wzrostu, który przyczynia się do hiperglikemii przez nasilenie glukoneogenezy i hamowanie zależnego od insuliny obwodowego poboru glukozy do komórek (IMGU) [5]. Rola cytokin polega na bezpośrednim wpływie na metabolizm glukozy oraz na stymulacji hormonów regulatorowych. Lang i wsp. zaobserwowali, że infuzja TNF α powoduje hyperglikemię, zwiększoną produkcję glukozy oraz oporność tkanek obwodowych na insulinę [53]. Z kolei Flores i wsp. doniesli, że TNF zwiększa osoczową glikemię przez stymulację glukoneogenezy wynikającą z pobudzenia sekrecji hormonów stresu [54]. Hiperglikemia będąca skutkiem działania cytokin wynika także z ich działania hamującego wydzielanie isnuliny [55]. Podczas reakcji stresowej głównymi substratami dla wątrobowej glukoneogenezy są mleczany i alanina [56]. U pacjentów z ostrym uszkodzeniem płuc (ALI), to właśnie płuca stanowią największe źródło mleczanów; inne źródła mleczanów podczas stresu to układ pokarmowy oraz rany [5,57]. Ważnym komórkowym producentem mleczanów są neutrofile oraz makrofagi. Infekcja oraz uraz powodują ich stymulację i nasilenie produkcji wolnych rodników tlenu, co jest związane ze zwiększonym przepływem glukozy przez szlak pentozofosforanowy i glikolizę, której produktem końcowym jest mleczan [58].
Reakcja stresowa powoduje zwiększenie uwalniania alaniny z mięśni szkieletowych, która następnie jest przekształcana w glukozę [59] oraz nasilenie lipolizy, której produktem jest glicerol także wykorzystywany w glukoneogenezie [5].
Ciężki uraz oraz infekcja są związane ze zwiększonym pobieraniem glukozy przez wszystkie tkanki organizmu. Jest to skutek nasilenia NIMGU przez cytokiny, który jest najwyraźniej zaznaczony w narządach zaangażowanych w odpowiedź zapalną na uraz lub infekcję (płuca, wątroba, śledziona i rany) [59,60,61,62]. Nasilenie NIMGU wynika ze zwiększonej syntezy, koncentracji błonowej i aktywności transportera GLUT -1 [63,64]. Reakcja stresowa jest stanem oporności na insulinę, gdyż hiperglikemii towarzyszy zazwyczaj podwyższone lub normalne osoczowe stężenie insuliny. Obwodowa oporność na insulinę wynika głównie z upośledzonego zależnego od insuliny poboru glukozy do komórek mięśni szkieletowych [36]. Odpowiadają za to cytokiny (TNF, IL -1, IL - 6),
które wpływają na szlak transdukcji sygnału receptorów dla insuliny zaburzając aktywację kinazy fosfatydyloinozytolu oraz na przemieszczanie się transporterów GLUT 4 do błony komórkowej [5, 65,66]. Rola katecholamin w rozwoju oporności na insulinę została zaobserwowana w badaniu, w którym blokada receptorów β2 u septycznych szczurów zapobiegała zmniejszeniu zależnego od insuliny poboru glukozy do komórek (IMGU) [67]. Nie jest znane dokładnie molekularne podłoże wpływu katecholamin na rozwój insulinooporności mięśni szkieletowych. W tkance tłuszczowej katecholaminy zmniejszają IMGU przez upośledzenie fosforylacji reszty tyrozynowej kinazy receptora dla insuliny [5].
Ośrodkowa insulinooporność może być definiowana jako zmniejszenie zdolności fizjologicznych stężeń insuliny do supresji zwiększonej wątrobowej produkcji glukozy [38]. Defekt ten może być jednak zniesiony przez duże dawki egzogennej insuliny [52].
Inne możliwe przyczyny hiperglikemii u pacjentów w ciężkim stanie to żywienia pozajelitowe [68], utajona lub wcześniejsza cukrzyca, marskość wątroby, zapalenie trzustki, leki (glukokortykoidy, diuretyki tiazydowe, fenytoina), hypokalemia, obniżenie poziomu chromu [5].
3.2.2. Niekorzystne następstwa hiperglikemii
Następstwa długotrwałej hiperglikemii u cukrzyków takie jak rozwój choroby niedokrwiennej serca, nefropatii cukrzycowej, retinopatii czy polineuropatii są powszechnie znane. Hiperglikemia towarzysząca ostrym stanom chorobowym także ma swoje negatywne konsekwencje. W 2005 roku przeprowadzono badanie na szczurach, u których indukowano hiperglikemię, następnie podawano endotoksynę i po 2 godzinach mierzono poziomy mediatorów zapalnych. W grupie hiperglikemicznych szczurów w porównaniu z kontrolą stwierdzono istotnie wyższe wartości mediatorów prozapalnych TNF α, Il -1, α1 kwaśnej glikoproteiny oraz kortykosteronu [69]. W badaniu z 2007 także zaobserwowano, iż hiperglikemia nasila stres oksydacyjny, produkcję czynników prozapalnych TNF α i Il-1, a z drugiej strony zmniejsza wytwarzanie antyoksydantów. Ponadto hiperglikemia jest uznawana za jedną z głównych przyczyn systemowej reakcji zapalnej SIRS [70]. Hiperglikemia powoduje zaburzenia gospodarki wodno -elektrolitowej indukowane przez glukozurię i odwodnienie organizmu. Podwyższony poziom glukozy w osoczu zaburza funkcję leukocytów, upośledzając adhezję granulocytów do komórek śródbłonka, upośledzając chemotaksję, fagocytozę, produkcję wolnych rodników, zabijanie wewnątrzkomórkowe oraz powoduje glikację układu dopełniacza zaburzając opsonizację [71]. Wszystkie te elementy osłabiające odpowiedź obronną organizmu sprzyjają zakażeniom czy to miejscowym czy
ogólnoustrojowym, które z większą częstością towarzyszą chorym hiperglikemicznym. Dodatkowo hiperglikemia nasila stres oksydacyjny, potencjalizuje cytokinową odpowiedź prozapalną, sprzyja reakcji prozakrzepowej i powoduje nieprawidłową odpowiedź naczyń krwionośnych na czynniki wazoaktywne, co w istotny sposób może zaburzać stan hemodynamiczny chorych z hiperglikemią [72]. U chorych z udarem niedokrwiennym serca zaobserwowano, iż hiperglikemia powoduje zwiększenie śmiertelności, zwłaszcza u pacjentów nie chorujących wcześniej na cukrzycę oraz przyczynia się do większego upośledzenia sprawności chorych po udarze niedokrwiennym [73,74]. Metaanaliza z 2000 roku dotycząca zaburzeń glikemii u chorych z ostrymi zespołami wieńcowymi wykazała, że pacjenci nie chorujący na cukrzycę z glikemią powyżej 110 mg/dl mieli 4 krotnie większe ryzyko zgonu w porównaniu z chorymi z niższymi wartościami glikemii. Stwierdzono także, iż glikemia przy przyjęciu powyżej 144 mg/dl u pacjentów nie chorujących wcześniej na cukrzycę wiązała się z większym ryzykiem rozwoju niewydolności serca oraz wstrząsu kardiogennego [5]. Z kolei badania nad zastosowaniem hormonu wzrostu u chorych oddziałów intensywnej terapii po zabiegach operacyjnych, urazach, z ostrą niewydolnością oddechową pokazały wzrost ryzyka zgonu o ponad 50 % w grupie chorych, u których stosowano hormon wzrostu. Autorzy tego badania dowodzą, że różnica w śmiertelności wynikała z wyższych wartości glikemii u chorych w grupie, w której stosowano hormon wzrostu [75].
4
Metabolizm glukozy w kontekście dysfunkcji
wybranych narządów
4.1
Choroby wątroby
Uraz czy krytyczna choroba powodują reakcję obronną organizmu z uruchomieniem kaskady cytokin i hormonów, które powodują zwiększoną wątrobową produkcję glukozy. Badania w grupie chorych oparzonych pokazały, iż produkcja glukozy wzrasta 1,5 krotnie w porównaniu do osób zdrowych [76]. Podczas gdy w warunkach fizjologicznych dzienna produkcja glukozy wynosi 200 g, chorzy oparzeni mogą zwiększyć produkcję glukozy do 320g/ dzień; wynika to ze zwiększonej glukoneogenezy [76]. Cewnikowanie naczyń nerkowych pokazało, że u chorych po urazie nie dochodzi do zwiększonej nerkowej produkcji glukozy w procesie glukoneogenezy, wobec czego wątroba odpowiada całkowicie za wzrost produkcji glukozy [76]. W związku z decydującą rolą wątroby w metabolizmie glukozy, choroby wątroby czy też zaburzenia jej funkcjonowania wynikające z uogólnionej infekcji powodują zaburzenia gospodarki węglowodanowej.
W badaniu z 2011 roku przedstawiającym metabolizm lipidów i węglowodanów u chorych z nie-alkoholowym stłuszczeniem wątroby (NASH), uznawanym za wątrobową manifestację zespołu metabolicznego, wykazano rozwój insulinooporności oraz upośledzenie testu doustnej tolerancji glukozy OGTT u pacjentów z wykładnikami NASH. Zaburzenia metabolizmu węglowodanów w tej jednostce chorobowej, prowadzącej do marskości wątroby, wynikają z uszkodzenia hepatocytów przez metabolity wolnych kwasów tłuszczowych, wolne rodniki tlenu oraz przez rozwój stanu zapalnego w obrębie komórek wątroby [77]. Zaburzenia glikemii oraz insulinooporność zaobserwowano także w badaniu z 2009 analizującym chorych z niealkoholową stłuszczeniową chorobą wątroby (NAFLD) oraz przewlekłym wirusowym zapaleniem wątroby typu C. Chorzy z NAFLD manifestują hiperglikemię oraz oporność na insulinę, których przyczyną mogą być nieprawidłowości gospodarki żelazem. Przeładowanie żelazem nasila wątrobowy stres oksydacyjny, który przyczynia się do włóknienia wątroby oraz zaburza przekazywanie sygnału od receptorów insuliny. Skutkiem zwłóknienia wątroby, hiperglikemii i hiperinsulinemii jest właśnie insulinooporność i zaburzenia poziomu glukozy w osoczu. Podobne nieprawidłowości dotyczą chorych z przewlekłym wirusowym zapaleniem wątroby typu C [78]. Badanie z 2011 roku dotyczące wpływu białka X wirusa HBV na glikemię u myszy zarażonych tym wirusem wykazało
zwiększoną ekspresją genów kodujących enzymy glukoneogenezy w wątrobie myszy zakażonych HBV. Stan zapalny wywołany infekcją wirusa HBV powoduje uwolnienie mediatorów zapalenia, TNF α oraz tlenku azotu, które powodują dysfunkcję wątroby, isnulinooporność i upośledzają działanie insuliny w obrębie wątroby [79]. W zmienionej zapalnie wątrobie pacjentów z przewlekłym wirusowym zapaleniem wątroby typu B wykazano zwiększoną ekspresją indukowalnej syntazy tlenku azotu [79], której poziom wzrasta także w przebiegu wstrząsu septycznego. W omawianym badaniu autorzy wykazali zwiększony poziom mRNA iNOS w wątrobie myszy zainfekowanych wirusem HBV, konkludujac iż hiperaktywność iNOS może odpowiadać za zwiększoną glukoneogenezę wątrobową zakażonych myszy. Badacze wykazali także, że pozbawienie części myszy ekspresji genu iNOS przyczyniło się do normalizacji poziomów glukozy [79]. Równocześnie stan zapalny w obrębie wątroby prowadzący do jej dysfunkcji jest wymieniany wśród czynników odpowiadających z występowanie epizodów hipoglikemii [80].
4.2
Choroby nerek
Wątroba oraz nerki są jedynymi narządami zdolnymi do produkcji glukozy w procesie glukoneogenezy. Enzym glukozo - 6 - fostataza obecny tylko w tych dwóch narządach powoduje odłączenie reszty fosforanowej od cząsteczki glukozy i uwolnienie wolnej glukozy do krążenia [5,32]. Pacjenci oddziałów intensywnej terapii mogą cierpieć na przewlekłą chorobę nerek jeszcze przed przyjęciem do oddziału intensywnej terapii lub mogą rozwinąć ostrą niewydolność nerek w przebiegu obecnej krytyczej choroby. Jako że nefropatia cukrzycowa stanowi najczęstszą przyczynę przewlekłej niewydolności nerek, zaburzenia glikemii wpisują się w obraz stanu klinicznego pacjentów z tym schorzeniem. Nadciśnienie tętnicze będące elementem zespołu metabolicznego, który charakteryzuje się insulinoopornością i wobec tego także zaburzeniami glikemii, stanowi drugą najczęstszą przyczyną przewlekłej choroby nerek. Nadciśnienie tętnicze ponadto, łącznie z hiperglikemią są niezależnymi czynnikami przyspieszającymi postęp przewlekłej niewydolności nerek. Z kolei wykazano, że przewlekła niewydolność nerek zwiększa ryzyko hiperglikemii [81,82]. Inne badanie analizujące najczęstsze przyczyny hipoglikemii, dowodzi, że niewydolność nerek stanowi drugie najczęstsze rozpoznanie u chorych z epizodem hipoglikemii (najczęstsze rozpoznanie stanowi cukrzyca) [80]. W badaniu z 2005 roku dokonującym analizy subpopulacji większego badania PICARD (Program to Improve Care in Acute Renal Disease), które zajmowało się chorymi oddziałów intensywnej terapii z ostrą niewydolnością nerek, wykazano, że w grupie
chorych, którzy przeżyli poziom glikemii był niższy, niż w grupie pacjentów, którzy zmarli. Związek między przeżyciem, a lepszą kontrolą glikemii był niezależny od wcześniejszej cukrzycy, wieku, płci, rasy, ciężkości choroby, poziomu kortyzolu w osoczu, stanu odżywienia, ciężkości ostrej niewydolności nerek definiowanej jako potrzeba terapii nerkozastępczej oraz sposobu żywienia. Stężenia insuliny były istotnie statystycznie wyższe w grupie chorych z ostrą niewydolnościa nerek, którzy zmarli w porównaniu z chorymi, którzy przeżyli. Analiza logistyczna mająca na celu określenie niezależnych czynników predykcyjnych zgonu chorych z ostrą niewydolnością nerek, uwzględniająca wiek, płeć, rasę, wynik w skali APACHE III, stężenie glukozy w okresie 5 tygodni, ciężkość ostrej niewydolności nerek (potrzebę terapii nerkozastępczej), stan odżywienia (poziom prealbumin) oraz sposób żywienia pacjentów, wykazała, że najlepszym czynnikiem predykcyjnym śmiertelność była ciężkość ostrej niewydolności nerek, stężenie glukozy, stężenie prealbumin i wynik w skali APACHE III. Autorzy badania wykazują także, iż u chorych z ostrą niewydolnością nerek dochodzi do rozwoju insulinooporności, stąd hiperglikemia i podwyższony poziom insuliny osoczowej u tych pacjentów. Konkludują, że insulinooporność może być czynnikiem wpływającym na śmiertelność u krytycznie chorych z ostrą niewydolnością nerek. Pacjenci w stanie krytycznym rozwijają insulinoporność w przebiegu ciężkiego stanu chorobowego, a ostra niewydolność nerek związana z utratą funkcji metabolicznej nerek może nasilać stan insulinooporności. W warunkach fizjologicznych insulina powoduje zmniejszenie nerkowej produkcj glukozy [83,84]. Brak natomiast doniesień na temat nerkowej produkcji glukozy u pacjentów z krytyczna chorobą. McGuiness i wsp. wykazali, że infuzja hormonów stresu psom powoduje wzrost nerkowej produkcji glukozy [81]. W stanie ostrej niewydolności nerek insulina traci jeden z istotnych organów docelowych swojego działania, wobec czego może pojawiać się insulinooporność. Podwyższony poziom mocznika w ostrej niewydolności nerek zmniejsza wątrobowe i obwodowe pobieranie glukozy przez komórki, także przyczyniając się do zaburzeń glikemii. W badaniach na szczurach, które miały podwyższony poziom mocznika wykazano zmniejszoną ilość transporterów dla glukozy w adipocytach [85]. Z drugiej strony hiperglikemia i hiperinsulinemia nasilając ostrą odpowiedź prozapalną i stres oksydacyjny mogą pogarszać rokowanie u chorych w stanie krytycznym z ostrą niewydolnością nerek [84,86,87].
4.3
Niewydolność nadnerczy związana z krytyczną chorobą -
Critical Illness Related Corticoid Insufficiency
W gospodarce węglowodanowej nieodzowną rolę odgrywają nadnercza i produkowane przez nie glikokortykosteroidy, które nasilają glukoneogenezę w wyniku zwiększonego katabolizmu białek, zwiększonego pobierania aminokwasów przez wątrobę, wzrostu aktywności enzymów glukoneogenezy; hamują one także zużywanie glukozy w tkankach pozawątrobowych, prowadząc do wzrostu poziomu glukozy osoczowej [32]. Jednym z istotnych elementów rozpoznania niewydolności nadnerczy, ostrej czy też przewlekłej jest hipoglikemia, wynikająca z braku hormonów kory nadnerczy [88]. Oprócz klasycznych form niewydolności nadnerczy, jak przykładowo choroba Addisona, rozpoznaje się też względną niewydolność nadnerczy (realtive adrenal insufficiency), która występować może u krytycznie chorych. W ostatnim czasie dużo uwagi poświęcano funkcji nadnerczy i terapii steroidowej u krytycznie chorych [89]. Stosowanie dużych dawek steroidów (10,000 do 40,000 mg hydrokortyzonu w ciągu doby) u chorych z ciężką sepsą i ARDS nie wiązało się z poprawą rokowania, wręcz przeciwnie skutkowało większą ilością zdarzeń niepożądanych [89,90]. Stosowanie dawek 200 do 350 mg na dobę przez okres 21 dni wiązało się z krótszym okresem wentylacji mechanicznej, krótszym pobytem w szpitalu oraz zmniejszyło śmiertelność w okresie kilku pierwszych dni pobytu w wybranych populacjach krytycznie chorych [91,92,93]. Chorzy, którzy odnieśli korzyści z terapii kortykosteroidami mieli nasiloną odpowiedź prozapalną, zwiększony poziom cytokin prozapalnych, takich jak TNF α, które odpowiadają za zmniejszoną syntezę glukokortykoidów przez hamowanie osi podwzgórze-przysadka-nadnercza, powodując względną niewydolność nadnerczy [89]. Dodatkowo u chorych w stanie krytycznym dochodzi do rozwoju oporności tkanek obwodowych na glikokortykosteroidy. Zjawisko to z angielskiego określane jako Critical Illness Related Corticosteroid Insufficiency (CIRCI), definiujemy jako nieadekwatną odpowiedź i aktywność glikokortykosteroidów w stosunku do ciężkości choroby. Manifestuje się niewystarczającą regulacją w dół czynników transkrypcyjnych mediatorów zapalenia, zależną od glikokortykoidów. Analogicznie do obrazu patofizjologicznego cukrzycy typu II (względny niedobór insuliny), w CIRCI dochodzi do oporność tkanek obwodowych na kortyzol oraz nieprawidłowego poziomu wolnego kortyzolu krążącego we krwi. W warunkach fizjologii ponad 90% kortyzolu krążącego we krwi związane jest z globuliną wiążącą kortyzol (CBG), a 10 % pozostaje w formie niezwiązanej [94]. W stanie choroby krytycznej, w szczególności w sepsie, poziom CBG obniża się o ponad 50 % i w konsekwencji zwiększa się poziom wolnego kortyzolu we krwi [95,96]. Oporność tkanek
obwodowych na kortyzol występuje w przebiegu takich chorób jak przewlekła obturacyjna choroba płuc, astma oskrzelowa, toczeń układowy, wrzodziejące zapalenie jelita grubego oraz reumatoidalne zapalenie stawów [89]. Podobnie w przypadku ostrego stanu zapalnego, jaki rozwija się w sepsie czy w ostrym uszkodzeniu płuc (ALI - acute lung injury), także dochodzi do oporności tkanek obwodowych na kortyzol [97]. Badania na modelu zwierzęcym ALI indukowanym toksyną E.coli wykazały zmniejszoną zdolność wiązania kortyzolu przez jego receptor jądrowy, która była przywracana do normy przez przeciwciała neutralizujące TNF i Il -1 [89]. Dodatkowo poza opornością tkanek na kortyzol, systemowej reakcji zapalnej towarzyszy niewydolność osi podwzgórze - przysadka - nadnercza, która występuje u około 20 % krytycznie chorych i u 60 % chorych z ciężką sepsą i wstrząsem septycznym [98]. Na niewystarczającą produkcję kortyzolu wpływa zmniejszona produkcja CRH, ACTH, zmiany strukturalne gruczołu nadnerczowego oraz niektóre leki, a także cytokiny prozapalne TNF i Il -1. TNF upośledza uwalnianie ACTH stymulowane przez CRH, a liczne badania dowodzą niskiego poziomu ACTH u chorych z ciężką sepsą [89,99]. TNF zmniejsza także syntezę kortyzolu hamując wpływ ACTH i angiotensyny II na nadnercza [89]. Zmniejszona produkcja kortyzolu może także wynikać z niedoborów substratów do produkcji tego hormonu, gdyż u krytycznie chorych, w tym chorych z sepsą, dochodzi do obniżenia poziomu cholesterolu HDL [100]. Manifestacja klinicza niewydolności nadnerczy towarzyszącej krytycznej chorobie jest konskekwencją nasilonej odpowiedzi prozapalnej organizmu. Częsty jest spadek ciśnienia tętniczego niereagujący na płynoterapię, wymagający włączenia amin katecholowych [101]. W badaniach laboratoryjnych u chorych z CIRCI występuje eozynofilia i hipoglikemia, rzadziej hiponatremia i hiperkalemia [89]. Zalecenia SSC z 2008 rekomendują stosowanie hydrokortyzonu dożylnie w dawce < 300mg/dobę we wstrząsie septycznym nie reagującym na płynoterapię i wazopresory.
4.4
Niewydolność trzustki
Pacjenci oddziałów intensywnej terapii najczęściej chorują na ostre zapalenie trzustki, które z jednej strony zaburza funkcję trzustki jako gruczołu dokrewnego, produkującego kluczowę hormony gospodarki węglowodanowej, a z drugiej strony uruchamia kaskadę cytokin prozapalnych wywołujących SIRS i prowadzących do hiperglikemii. Hiperglikemia, która stosunkowo często występuje w przebiegu ostrego zapalenia trzustki, jest związana ze zwiększonym ryzykiem powikłań infekcyjnych i zwiększoną śmiertelnością [102]. W badaniach oceniających funkcję trzustki i homeostazę glikemiczną u chorych, którzy przebyli ostre zapalenie trzustki, wykazano, iż
u części chorych dochodzi do zaburzeń przemiany glukozy [103,104]. Szczególnie dotyczy to chorych z ciężką postacią ostrego zapalenia trzustki, którzy w badaniu z 2010 roku mieli wyższe wartości glikemii na czczo, wyższe wartości glikemii w 120 minucie doustnego testu obciążenia glukozą. Prawie 80 % chorych z ciężką postacią ostrego zapalenia trzustki spełniało kryteria rozpoznania cukrzycy oraz upośledzonej tolerancji glukozy. Chorzy, którzy rozwinęli cukrzycę mieli wyższe wartości peptydu C na czczo oraz wyższe poziomy insuliny wskazujące na insulinooporność. Z drugiej strony chorzy ze zmianami martwiczymi trzustki mają zmniejszoną liczbę komórek β i wobec tego mieli niższe poziomy peptydu C i insuliny, co także może wpływać na hiperglikemię u tych chorych [103].
5
Badania własne
Rekomendacje autorów wytycznych Surviving Sepsis Campaign zalecające kontynuowanie badań nad ustaleniem optymalnego poziomu glikemii u chorych z sepsą oraz burzliwe rozważania w literaturze ostatnich 10 lat dotyczące intensywnej insulinoterapii i kontroli glikemii u chorych oddziałów intensywnej terapii stały się fundamentem do zanalizowania profilu glikemicznego oraz schematu insulinoterapii stosowanego w oddziale intensywnej terapii poznańskiego szpitala klinicznego.
5.1
Hipotezy badawcze
Postawione zostały następujące hipotezy badawcze:
1. Pacjenci, którzy zmarli z powodu ciężkiej sepsy lub wstrząsu septycznego mieli częściej epizody hiperglikemii i hipoglikemii.
2. Pacjenci, którzy zmarli z powodu ciężkiej sepsy lub wstrząsu septycznego mieli większe dobowe wahania glikemii.
3. Funkcja innych narządów oraz interwencje terapeutyczne wpływają na profil glikemii u chorych z ciężką sepsą oraz wstrząsem septycznym.
5.2
Materiał i metoda badań
Materiał badawczy stanowiła dokumentacja medyczna 98 chorych Kliniki Anestezjologii, Intensywnej Terapii i Leczenia Bólu Szpitala Klinicznego im. Heliodora Święcickiego w Poznaniu przyjętych do oddziału w latach 2005 - 2009 z rozpoznaniem ciężkiej sepsy i wstrząsu septycznego klasyfikowanych zgodnie z zaleceniami American College of Chest Physicians i Society of Critical Care Medicine z 1992 roku, konferencji uzgodnieniowej International Sepsis Definitions Conference w 2001 oraz rekomendacjami Surviving Sepsis Campaign z 2008 roku. Retrospektywnej analizie poddano profil glikemiczny pacjentów z sepsą w szerokim kontekście uwzględniającym choroby współistniejące, dysfunkcję kluczowych narządów oraz interwencje terapeutyczne. Badaniem objęto okres pierwszych czternastu dni pobytu w oddziale intensywnej terapii, który charakteryzował się najbardziej intensywnym postępowaniem diagnostyczno-terapeutycznym.
Schemat insulinoterapii stosowany w oddziale polega na dożylnym ciągłym wlewie ludzkiej insuliny krótkodziałającej przy pomocy pomp infuzyjnych według ustalonego schematu przedstawionego w tabeli poniżej.
Stężenie glukozy (mg/dl) Szybkość wlewu insuliny (IU/h)
0 - 80 0 80 - 125 1 126 - 180 2 181 - 235 3 236 - 305 4 306 - 380 5 > 380 wezwij lekarza
Schemat ten nadzorowany jest przez zespół pielęgniarski, który dostosowuje szybkość wlewu insuliny do wartości aktualnie zmierzonej glikemii zgodnie ze schematem z tabeli. Standardowo co sześć godzin pobierana jest próbka krwi tętniczej, wykonywana jest gazometria i poziom glikemii. Docelowa wartość glikemii mieści się w przedziale 80 - 150 mg/dl. Gdy poziom glikemii wykracza poza rekomendowany przedział pomiar glikemii wykonywany jest co 1 lub 2 godziny, do czasu powrotu wartości glikemii do zalecanego poziomu. Lekarz jest informowany, gdy glikemia spada
poniżej 80 mg/dl lub gdy przekracza wartość 380 mg/dl. Analizie lekarskiej poddawany jest także całodobowy profil glikemiczny danego pacjenta. Decyzje terapeutyczne podejmowane są indywidualnie dla każdego pacjenta. W przypadku hipoglikemii podawany jest z reguły dożylnie bolus 10 ml 20% lub 40 % glukozy. Z kolei w przypadku wysokich wartości glikemii przykładowo zmniejszana jest szybkość podaży żywienia pozajelitowego lub wprowadza się do diety enteralnej jednonienasycone kwasy tłuszczowe jako główne źródło energii.
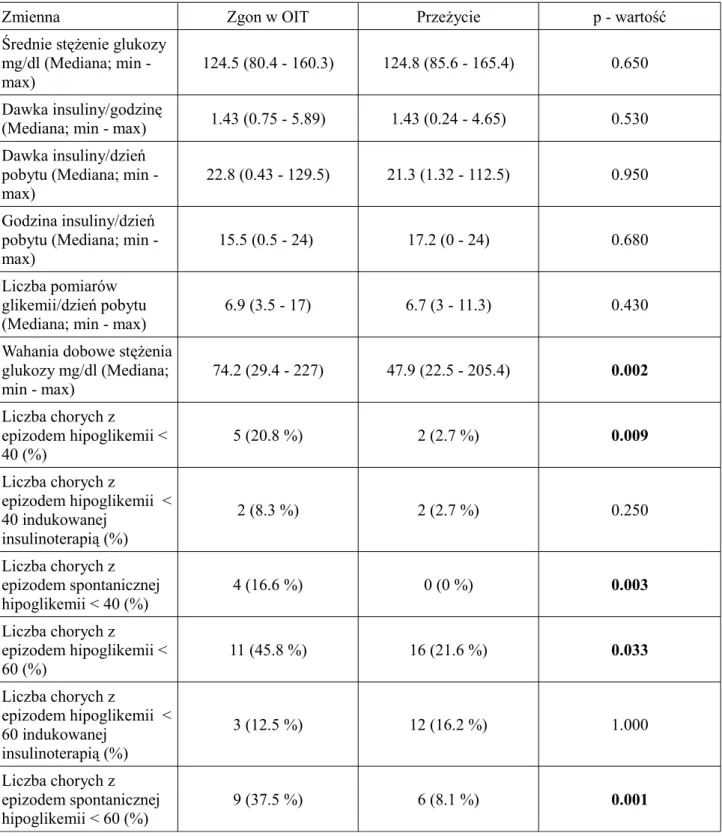
Analiza profilu glikemicznego chorych z ciężką sepsą oraz wstrząsem septycznym obejmowała średnie poziomy glikemii, dobowe wahania glikemii (określane jako różnica między maksymalną a minimalną wartością glikemii w ciągu doby), czas insulinoterapii, średnie dawki insuliny w przeliczeniu na godzinę oraz na dobę, epizody hipoglikemii < 40 mg/dl - określane w piśmiennictwie jako ciężka hipoglikemia, hipoglikemii < 60 mg/dl - określane jako wartość graniczna glikemii dla funkcjonowania komórek ośrodkowego układu nerwowego, hipoglikemii spontanicznej < 40 mg/dl i < 60 mg/dl - definiowanej jako hipoglikemia, gdy ciągły dożylny wlew insuliny przez pompę infuzyjną był wstrzymany przynajmniej na 4 godziny oraz hipoglikemii < 40 i < 60 mg/dl występujące w trakcie dożylnego wlewu insuliny. Ponadto analizowano epizody hipoglikemii < 80 mg/dl - jest to górna wartość przedziału, w którym według schematu insulinoterapii stosowanego w oddziale nie należy podawać wlewu insuliny; hiperglikemii > 150 mg/dl - gdzie wartość 150 mg/dl stanowi górną granicę wartości glikemii zalecanych u chorych z sepsą, hiperglikemii > 180 mg/dl - granicznej wartości glikemii zalecanej w badaniu NICE - SUGAR oraz epizodów hiperglikemii > 200 mg/dl, która to wartość stanowi jedno z kryteriów rozpoznania cukrzycy. W analizie uwzględniono choroby współistniejące (cukrzyca, ostra lub przewlekła niewydolność nerek, ostre zapalenie trzustki, choroba niedokrwienna serca, nadciśnienie tętnicze, choroba nowotworowa w wywiadzie), wybrane parametry laboratoryjne (gazometrię tętniczą, poziom białka C reaktywnego - CRP, mleczanów, elektrolitów, parametry morfologii, funkcji nerek i wątroby), leki (aminy katecholowe, steroidy) i działania terapeutyczne (stosowanie żywienia pozajelitowego, hemodializoterapia, ciągła żylno-żylna hemofiltracja - CVVH ) mogące wpłynąć na profil glikemiczny. Ocenę profilu glikemicznego przeprowadzono porównując grupę chorych, którzy zmarli w oddziale intensywnej terapii i którzy przeżyli wypisani do innego oddziału oraz grupę chorych, którzy zmarli w szpitalu w trakcie analizowanej hospitalizacji z grupą chorych, którzy zostali wypisani ze szpitala. Pozostałe analizy porównawcze uwzględniały jako zmienną grupującą cukrzycę rozpoznaną przed przyjęciem do oddziału intensywnej terapii, ostre zapalenie trzustki, wystąpienie epizodu hipoglikemii < 60 mg/dl, epizodu hiperglikemii > 180 mg/dl, stosowanie terapii nerkozastępczej, steroidów, żywienia pozajelitowego, noradrenaliny, poziom wskaźnika INR > 1.5, poziom bilirubiny > 1.0 mg/dl odzwierciedlające fukcję wątroby oraz poziom
kreatyniny > 1.2 mg/dl odzwierciedlający funkcję nerek.
Analiza statystyczna została przeprowadzona w oparciu o program komputerowy Statistica edycja 8. Zebrane dane zostały podzielone względem skali interwałowej i nominalnej, w oparciu o te skale dokonano analizy statystycznej bazującej na porównywaniu dwóch grup względem wybranego parametru, takiego jak przykładowo zgon bądź przeżycie w oddziale intensywnej terapii, wystąpienie epizodu hipoglikemii < 60 mg/dl. Ponadto dokonano oceny czynników ryzyka wystąpienia określonych zdarzeń, przeprowadzając obliczenia współczynnika odds ratio, czyli ilorazu szans. Analizy ilorazu szans dokonano dla tych parametrów, które okazały się istotne statystycznie. Za poziom istotności statystycznej przyjęto p - wartość równą 0,05.W pierwszym etapie sprawdzano wystąpienie rozkładu normalnego danych w skali interwałowej stosując test Shapiro-Wilka. Dane, które spełniały warunki normalność analizowano używając testu t-studenta, korzystając z obliczenia parametrów tesut Levene'a. Gdy p- wartość testu Levene'a wynosiła powyżej 0,05 korzystano z testu t-studenta, natomiast gdy p - wartość testu Levene'a wynosiła poniżej 0,05 korzystana z testu z niezależną estymacją wariancji - testu Welcha. Przedstawiając dane w skali interwałowej, które spełniały warunek normalności stosowano wartości średnie i odchylenie standardowe danego parametru. Dane, które nie spełniały warunku rozkładu normalnego analizowano testami nieparametrycznymi, bazując na teści U Manna - Whitneya. W odniesieniu do danych nie spełniających kryteriów normalności stosowano medianę i wartości minimalne i maksymalne Z kolei dane w skali nominalnej, takie jak przykładowo wystąpienie epizodu hiperglikemii > 200 mg/dl bądź obecność choroby nowotworowej w wywiadzie analizowano korzystając z tabeli porównawczej 2 x 2 i dokładnego testu Fishera dwustronnego. Dane w skali nominalnej przedstawiono w postaci wartości liczbowych i wartości procentowych.
5.3
Wyniki
Oceny profilu glikemicznego chorych septycznych dokonano porównując wybrane parametry demograficzne, kliniczne oraz laboratoryjne względem zmiennych grupujących, takich jak zgon w oddziale intensywnej terapii, zgon szpitalny, wystąpienie epizodu hipoglikemii < 60 mg/dl, wystąpienie epizodu hiperglikemii > 180 mg/dl, stosowanie terapii nerkozastępczej CRRT (continous renal replecament therapy), żywienia pozajelitowego, noradrenaliny, choroba trzustki, cukrzyca, poziom INR > 1.5 , poziom bilirubiny > 1 mg/dl, poziom kreatyniny > 1.2 mg/dl.
1. Porównanie chorych z ciężką sepsą i wstrząsem septycznym, którzy zmarli w oddziale intensywnej terapii z chorymi, którzy przeżyli
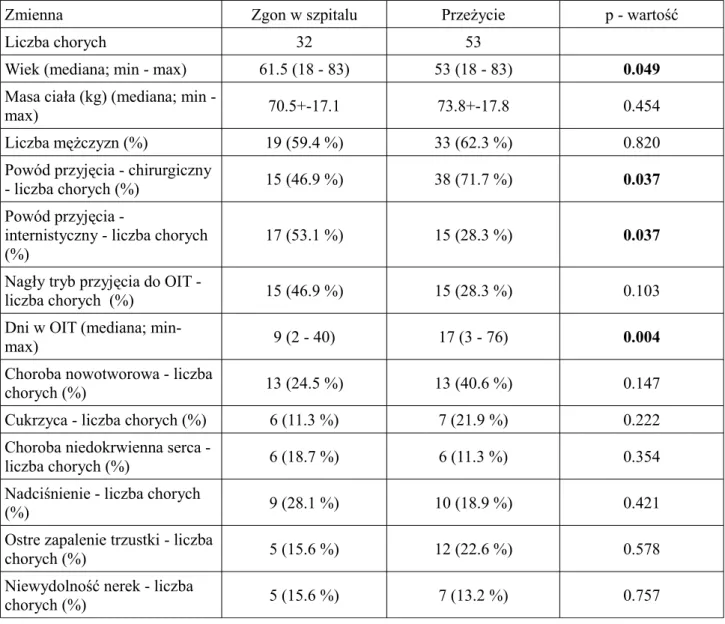
Tabela 1. Dane demograficzne i kliniczne
Zmienna Grupa 1 – Zgon w OIT Grupa 2 – Przeżycie p - wartość
Liczba chorych 24 74
Wiek (mediana; min - max) 59 (18 - 76) 53 (18 - 83) 0.170 Masa ciała (kg) (mediana; min -
max) 67,5 (28 - 98) 72,5 (37 - 160) 0.790
Liczba mężczyzn (%) 14 (58.3 %) 47 (63.5 %) 0.800
Powód przyjęcia chirurgiczny -
liczba chorych (%) 8 (33.3 %) 52 (70.3 %) 0.001
Powód przyjęcia internistyczny
- liczba chorych (%) 16 (66.6 %) 22 (29.7 %) 0.001
Nagły tryb przyjęcia do OIT -
liczba chorych (%) 14 (58.3 %) 22 (29.7 %) 0.015
Dni w OIT (mediana; min-
max) 9 (2 - 40) 14 (1 - 75) 0.018
Choroba nowotworowa - liczba
chorych (%) 7 (29.2 %) 22 (29.7 %) 1.000
Cukrzyca - liczba chorych (%) 5 (20.8 %) 10 (13.5 %) 0.510 Choroba niedokrwienna serca -
liczba chorych (%) 5 (20.8 %) 9 (12.2 %) 0.320
Nadciśnienie - liczba chorych
(%) 6 (25 %) 15 (20.3 %) 0.770
Ostre zapalenie trzustki - liczba
chorych (%) 5 (20.8 %) 14 (18.9 %) 1.000
Niewydolność nerek - liczba
Nie wykazano istotnych statystycznie różnic między chorymi, którzy zmarli w oddziale intemsywnej terapii, a chorymi którzy przeżyli podwzględem danych demograficznych. Podobnie w odniesieniu do chorób współistniejących porównywalny odsetek chorych w obu analizowanych grupach miał rozpoznane ostre zapalenie trzustki, cukrzycę, nadciśnienie tętnicze, chorobę niedokrwienną serca, chorobę nowotworową oraz ostrą niewydolność nerek. Zaobserwowano, iż chorzy z ciężką sepsą i wstrząsem septycznym, którzy zmarli w oddziale intensywnej terapii byli przyjmowani do oddziału w trybie nagłym oraz z powodów tzw. internistycznych, z angielskiego medical.
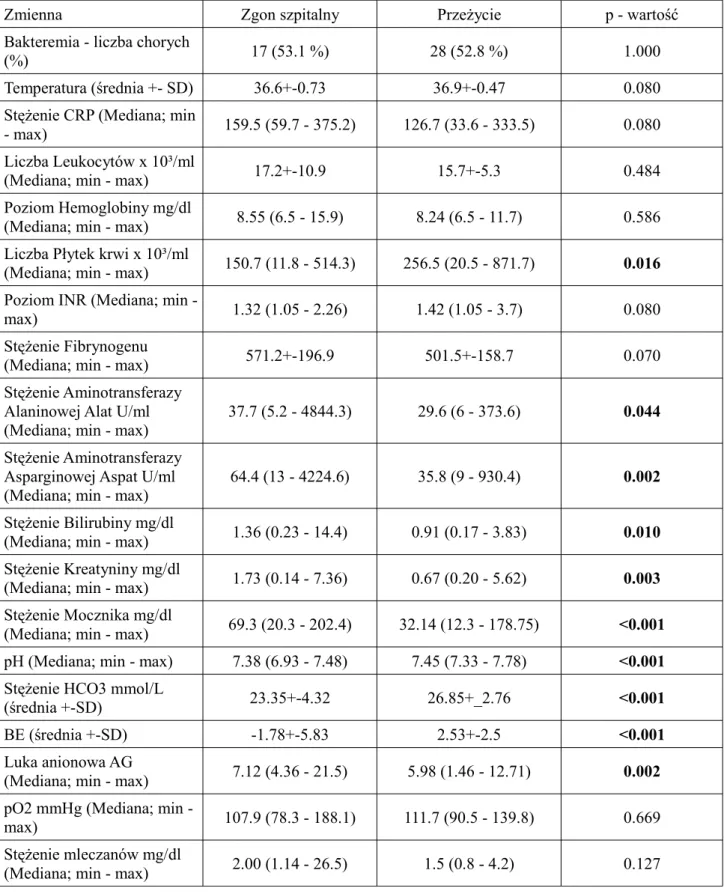
Tabela 2. Mierzone wielkości laboratoryjne - część 1
Zmienna Zgon w OIT Przeżycie p - wartość
Bakteremia - liczba chorych (%) 13 (54.2 %) 38 (51.3 %) 1.000 Temperatura (średnia +- SD) 36.6 +- 0.8 36.8+-0.5 0.210 Stężenie CRP (Mediana; min - max) 169.6 (59.7 - 375.2) 122.7 (33.6 - 333.5) 0.023 Liczba Leukocytów x 10³/ml (Mediana; min - max) 15.7 (0.3 - 42.2) 14.6 (6.6 - 44.2) 0.596 Poziom Hemoglobiny mg/dl (Mediana; min - max) 8.27 (6.5 - 15.8) 8.5 (6.5 - 11.8) 0.210
Liczba Płytek krwi x 10³/ml (Mediana; min - max)
170.2 (11.8 - 514.3) 212.5 (20.5 - 871.1) 0.090
Poziom INR (Mediana;
min - max) 1.37 (1.05 - 2.26) 1.43 (1.05 - 3.7)
0.460
Stężenie Fibrynogenu
(Mediana; min - max) 572.7 (102 - 790.8) 512.9 (102 - 1058) 0.250 Stężenie Aminotransferazy
Alaninowej Alat U/ml (Mediana; min - max)
54.5 (5.2 - 4884) 28 (6 - 373.6) 0.006
Stężenie Aminotransferazy Asparginowej Aspat U/ml (Mediana; min - max)
90.5 (13 - 4224) 35.5 (9 - 930) <0.001
Stężenie Bilirubiny mg/dl
Tabela 3. Mierzone wielkości laboratoryjne - część 2
Zmienna Zgon w OIT Przeżycie p - wartość
Stężenie Mocznika mg/dl (Mediana; min - max)
72.9 (33.7 - 202.4) 36.6 (12.3 - 178.7) <0.001
Stężenie Kreatyniny mg/dl (Mediana; min - max)
2.04 (0.22 - 7.36) 0.71 (0.14 - 5.6) 0.001
pH (Mediana; min - max) 7.37 (6.93 - 7.48) 7.45 (7.3 – 7.78) <0.001
Stężenie HCO3 mmol/L (średnia +-SD)
22.7+- 4.6 26.8+-2.6 <0.001
BE średnia +-SD) -2.7 +-6.3 2.5+-2.5 <0.001
Luka anionowa AG (Mediana; min - max)
9.26 (4.36 - 21.5) 6.07 (0.7 - 14) <0.001 pO2 mmHg (Mediana; min - max) 107.4 (78.3 - 188) 111.0 (90.5 - 152.8) 0.150 Stężenie mleczanów mg/dl (Mediana; min - max) 3.57 (1.14 - 26.4) 1.5 (0.8 - 23.3) 0.033
Analiza danych laboratoryjnych wykazała, iż chorzy którzy zmarli w oddziale intensywnej terapii mieli statystycznie istotnie wyższe wartości parametrów funckji wątroby - Alat, Aspat, bilirubina oraz funkcji nerek - kreatynina oraz mocznik. Podobnie chorzy ci mieli zdecydowanie gorsze parametry gospodarki kwasowo-zasadowej, takie jak pH, HCO3, BE, luka anionowa, stężenie mleczanów, z wyjątkiem prężności tlenu pO2. W odniesieniu do markerów stanu zapalnego chorzy, którzy zmarli w oddziale intensywnej terapii mieli wyższe wartości CRP. Zależności takiej nie wykazano dla temperatury oraz ilości leukocytów. Porównywalne między obu grupami były wartości morfologii krwi, takie jak stężenie hemoglobiny, liczba płytek krwi oraz parametry układu krzepnięcia - INR oraz stężenie fibrynogenu.
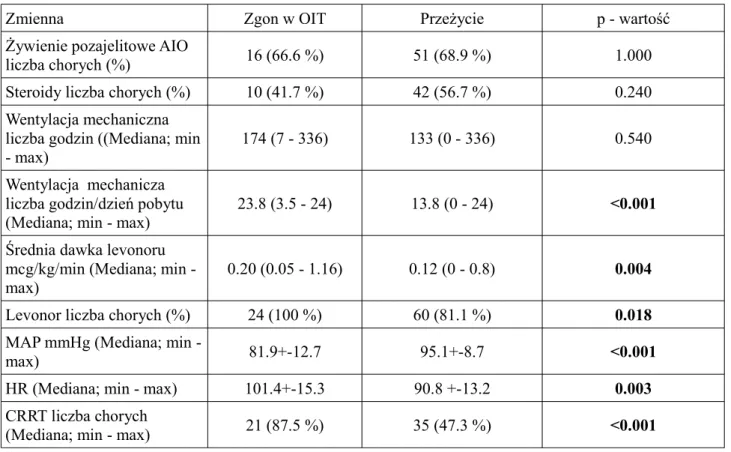
Tabela 4. Zmienne związane ze stosowaną terapią
Zmienna Zgon w OIT Przeżycie p - wartość
Żywienie pozajelitowe AIO
liczba chorych (%) 16 (66.6 %) 51 (68.9 %) 1.000
Steroidy liczba chorych (%) 10 (41.7 %) 42 (56.7 %) 0.240 Wentylacja mechaniczna
liczba godzin ((Mediana; min - max)
174 (7 - 336) 133 (0 - 336) 0.540
Wentylacja mechanicza liczba godzin/dzień pobytu (Mediana; min - max)
23.8 (3.5 - 24) 13.8 (0 - 24) <0.001
Średnia dawka levonoru mcg/kg/min (Mediana; min - max)
0.20 (0.05 - 1.16) 0.12 (0 - 0.8) 0.004
Levonor liczba chorych (%) 24 (100 %) 60 (81.1 %) 0.018
MAP mmHg (Mediana; min -
max) 81.9+-12.7 95.1+-8.7 <0.001
HR (Mediana; min - max) 101.4+-15.3 90.8 +-13.2 0.003
CRRT liczba chorych
(Mediana; min - max) 21 (87.5 %) 35 (47.3 %) <0.001
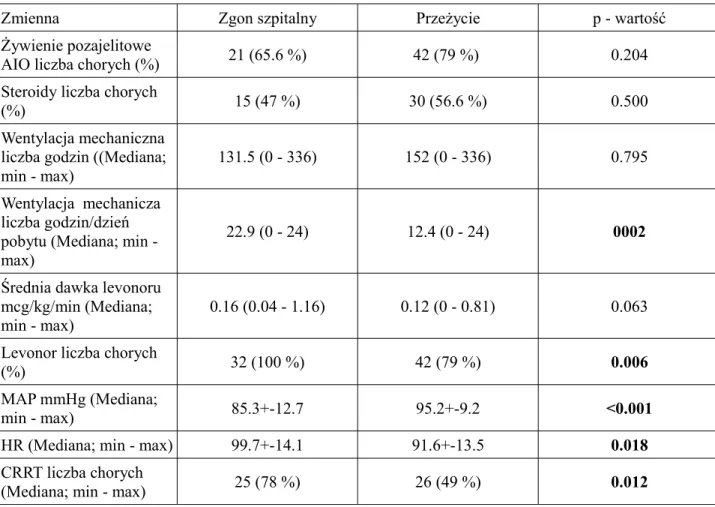
W odniesieniu do elementów terapii chorych z ciężką sepsą oraz wstrząsem septycznym nie wykazano różnicy w odsetku chorych w obu grupach, którzy wymagali żywienia pozajelitowego oraz steroidoterapii, a także w sumarycznej liczbie godzin wentylacji mechanicznej. Zaobserwowano natomiast, iż chorzy, którzy zmarli w oddziale intensywnej terapii wymagali większych dawek levonoru oraz większej liczby godzin wentylacji mechanicznej w przeliczeniu na dzień pobytu. Chorzy ci mieli także istotnie statystycznie niższe wartości średniego ciśnienia tętniczego oraz większą częstość pracy serca. Podobnie większy odsetek chorych, którzy nie przeżyli wymagał stosowania amin presyjnych oraz terapii nerkozastępczej.
Tabela 5. Profil glikemiczny - część 1
Zmienna Zgon w OIT Przeżycie p - wartość
Średnie stężenie glukozy mg/dl (Mediana; min - max)
124.5 (80.4 - 160.3) 124.8 (85.6 - 165.4) 0.650
Dawka insuliny/godzinę
(Mediana; min - max) 1.43 (0.75 - 5.89) 1.43 (0.24 - 4.65) 0.530 Dawka insuliny/dzień
pobytu (Mediana; min - max)
22.8 (0.43 - 129.5) 21.3 (1.32 - 112.5) 0.950
Godzina insuliny/dzień pobytu (Mediana; min - max)
15.5 (0.5 - 24) 17.2 (0 - 24) 0.680
Liczba pomiarów glikemii/dzień pobytu (Mediana; min - max)
6.9 (3.5 - 17) 6.7 (3 - 11.3) 0.430
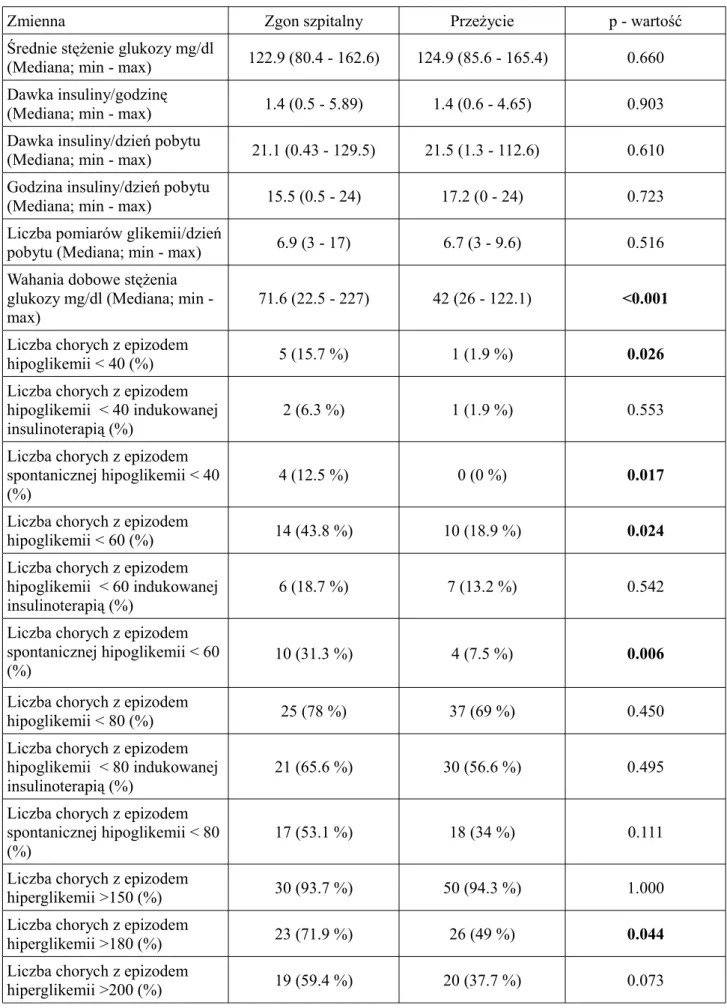
Wahania dobowe stężenia glukozy mg/dl (Mediana; min - max) 74.2 (29.4 - 227) 47.9 (22.5 - 205.4) 0.002 Liczba chorych z epizodem hipoglikemii < 40 (%) 5 (20.8 %) 2 (2.7 %) 0.009 Liczba chorych z epizodem hipoglikemii < 40 indukowanej insulinoterapią (%) 2 (8.3 %) 2 (2.7 %) 0.250 Liczba chorych z epizodem spontanicznej hipoglikemii < 40 (%) 4 (16.6 %) 0 (0 %) 0.003 Liczba chorych z epizodem hipoglikemii < 60 (%) 11 (45.8 %) 16 (21.6 %) 0.033 Liczba chorych z epizodem hipoglikemii < 60 indukowanej insulinoterapią (%) 3 (12.5 %) 12 (16.2 %) 1.000 Liczba chorych z epizodem spontanicznej hipoglikemii < 60 (%) 9 (37.5 %) 6 (8.1 %) 0.001