Uniwersytet im. Adama Mickiewicza w Poznaniu
Wydzia Chemii
Zak ad Technologii Chemicznej
Skrypt przeznaczony dla studentów studiów stacjonarnych
IV roku chemii,
chemii rodowiska,
chemii materia owej
i zawodowego studium chemii
Zespó autorów: Maria
Wojciechowska
Mariusz Pietrowski
Micha Zieli ski
SPIS TRE+CI
1. Wst-p ... 7 2. Spis /wicze ... 9 3. Opisy /wicze
3wiczenie 1. Cia o sta e w procesie katalitycznej redukcji NO
w-glowodorami ... 11 3wiczenie 2. Zastosowanie nowych materia ów jako katalizatorów
do usuwania NO przy pomocy tlenku w-gla ... 29 3wiczenie 3. Wykorzystanie cia sta ych w badaniach mechanizmów
reakcji katalitycznych. Mechanizm redukcji NO propenem ... 39 3wiczenie 4. Temperaturowo programowana redukcja wodorem (TPR-H2)
w badaniach oddzia ywa faza aktywna-no<nik ... 45 3wiczenie 5. Termodesorpcja amoniaku (TPD-NH3) jako metoda
wyznaczania kwasowo<ci cia sta ych ... 58 3wiczenie 6 Badanie kwasowo<ci cia a sta ego metodA FTIR
z uCyciem czAsteczek sond ... 65 3wiczenie 7. Identyfikacja centrów aktywnych na powierzchni
cia a sta ego metodA reakcji modelowych ... 77 3wiczenie 8. Wp yw rodzaju warstwy przypowierzchniowej
na w a<ciwo<ci chemiczne danego uk adu ... 89 4. Regulamin pracowni ... 101 Telefony alarmowe ... 102 5. Podstawowe zasady bezpiecze stwa i pracy
„Chemia Cia a Sta ego. wiczenia Laboratoryjne” s przeznaczone dla studentów studiów stacjonarnych IV roku chemii, chemii %rodowiska, chemii materia owej i zawodowego studium chemii odbywaj cych kurs chemii na Wydziale Chemii Uniwersytetu im. A. Mickiewicza w Poznaniu. Materia y te zawieraj opis +wicze, oraz regulamin pracowni i opis podstawowych zasad organizacji bezpiecznej pracy laboratoryjnej.
Zestaw opisów +wicze, laboratoryjnych jest uzupe niony podstawowymi wiadomo%ciami teoretycznymi obrazuj cymi poszczególne techniki wykorzystywane w badaniach cia sta ych. Znajomo%+ tych wiadomo%ci jest konieczna do wykonania +wiczenia.
Celem +wicze, z chemii cia a sta ego jest zapoznanie studenta z podstawowymi technikami pozwalaj cymi scharakteryzowa+ struktur. i chemi. powierzchni cia a sta ego oraz niektóre w a%ciwo%ci katalityczne nowych materia ów. Ogólna koncepcja realizacji tego celu uwzgl.dnia zatem zarówno zapoznanie si. studentów z podstawami teoretycznymi jak i praktyczne przeprowadzenie eksperymentów chemicznych.
Prof. dr hab. Maria Wojciechowska Kierownik Zak adu Technologii Chemicznej
WSTEP
SPIS 3WICZEF
3wiczenie 1. Cia o sta e w procesie katalitycznej redukcji NO w-glowodorami
3wiczenie 2. Zastosowanie nowych materia ów jako katalizatorów do usuwania NO przy pomocy tlenku w-gla
3wiczenie 3. Wykorzystanie cia sta ych w badaniach mechanizmów reakcji katalitycznych. Mechanizm redukcji NO propenem
3wiczenie 4. Temperaturowo programowana redukcja wodorem (TPR-H2)
w badaniach oddzia ywa faza aktywna-no<nik
3wiczenie 5. Termodesorpcja amoniaku (TPD-NH3) jako metoda wyznaczania
kwasowo<ci cia sta ych
3wiczenie 6. Badanie kwasowo<ci cia a sta ego metodA FTIR z uCyciem czAsteczek sond
3wiczenie 7. Identyfikacja centrów aktywnych na powierzchni cia a sta ego metodA reakcji modelowych
3wiczenie 8. Wp yw rodzaju warstwy przypowierzchniowej na w a<ciwo<ci chemiczne danego uk adu
1
Cia o sta e w procesie katalitycznej
redukcji NO w-glowodorami
Literatura uzupe niaj ca:
Peter O’Neill, „Chemia %rodowiska” Wydawnictwo Naukowe PWN, Warszawa-Wroc aw 1998, str. 117-138.
Maria Zió ek, Izabela Nowak, „Kataliza Heterogeniczna – wybrane zagadnienia”, Wydawnictwo Naukowe UAM, Pozna4 1999, str. 118-122.
1. Wst-p
„Wielko</ dawki czyni trucizn-” – s owa Paracelsusa. Znaczy to, 1e toksyczne dzia anie danej substancji zale1y od dawki i 1e poni1ej pewnych ilo%ci nie ma ona truj cego dzia ania.
Tej podstawowej prawdzie cz.sto przeciwstawia si. tak zwan hipotez- liniowA która zak ada, 1e substancja truj ca w du1ych dawkach musi by+ truj ca tak1e w dawkach ma ych. Hipoteza ta nie jest jednak zgodna ze zdrowym rozs dkiem, co mo1na wykaza+ na przyk adzie tlenku azotu.
Otó1 tlenek azotu jest krótko 1yj c cz steczk – sk adaj c si. z dwu atomów, dwóch najpospolitszych na ziemi gazów, tj. tlenu i azotu. Jest on bezbarwnym i bezwonnym gazem. Do niedawna wiadomo by o w a%ciwie tyle, 1e znajduj si. w spalinach samochodowych i 1e wchodzi w reakcj. z innymi sk adnikami atmosfery, przyczyniaj c si. do powstania smogu. Dopiero od 1987 roku wydarzenia zwi zane z tlenkiem azotu okaza y si. wr.cz sensacyjne. W 1992 roku w czasopi%mie „Science” uznano NO za „cz steczk. roku”. W 1998 roku trzech uczonych uzyska o nagrod. Nobla za prace nad korzystnym wp ywem NO na nasz organizm.
2. Pozytywna rola NO
Obecnie wiadomo, 1e tlenek azotu w ma ych ilo%ciach pe ni bardzo wa1ne funkcje biologiczne, jest niezb.dny do prawid owego funkcjonowania serca, tj. w ilo%ci 0,6-2,1
µmol/litr (tworzy si. w trakcie skurczów serca). Wi.ksze ilo%ci NO powoduj zbytnie
rozszerzanie naczy, i serce nie nad 1a pompowa+ krwi. Pe ni on równie1 bardzo wa1ne funkcje biologiczne np.:
1. W uk adzie nerwowym jest neurotransmiterem.
2. Pe ni funkcje obronne w organizmie – chroni przed paso1ytami, bakteriami, by+ mo1e przed wirusami.
3. Jest czynnikiem kontroluj cym ci%nienie krwi.
4. Zapobiega przyleganiu krwinek do %ciany i powstawaniu blaszki mia1d1ycowej.
NO znalaz równie1 wykorzystanie w leczeniu wielu chorób. Kuracja tlenkiem azotu pomaga prze1y+ noworodkom, u których wyst pi y problemy z oddychaniem. Sta o si. równie1 jasne, dlaczego stosowane od lat leki, np. nitrogliceryna, która rozk ada si. do NO, s skuteczne w leczeniu chorób uk adu kr 1enia.
3. Negatywnie dzia ajAce tlenki azotu; ich pochodzenie
i sposoby ograniczenia
Pozytywna rola NO zwi zana jest z jego %ci%le okre%lon dawk . Jej przekroczenie mo1e doprowadzi+ do utraty przytomno%ci, a w konsekwencji nawet do %mierci. I tak epidemiolodzy podaj , 1e przebywanie przez 24 godzin w atmosferze zawieraj cej NO w ilo%ci 0,005 ppm negatywnie wp ywa na nasz organizm. Poziom ten cz.sto jest rejestrowany w obszarach uprzemys owionych i g.sto zaludnionych.
Za emisj. tlenków azotu w tych obszarach odpowiedzialne s gazy spalinowe pochodz ce ze spalania paliw naturalnych w elektrowniach zak adach przemys owych, czy
technologie przemys owe 30% m ieszkalnic-tw o 32% m otoryzacja 29% energetyka 9%
produktów ropy naftowej szczególnie w pojazdach samochodowych.
W%ród emitowanych tlenków azotu (NOx) najwi.kszy udzia stanowi NO ~90-95%.
Udzia tych ostatnich jest szczególnie du1y w krajach uprzemys owionych i mo1e wynosi+ nawet ~60% ca kowitej emisji tlenków azotu. Spora cz.%+ zanieczyszcze, tlenkami azotu jest tak1e pochodzenia naturalnego. Tworz si. w trakcie spalania lasów, wybuchów
wulkanów, burz piaskowych, huraganów czy procesów rozk adu materii organicznej. Obraz emitowanych tlenków azotu w Polsce przedstawia si. nast.puj co (rys. 1).
Tlenek „wytwarzany” przez cz owieka powstaje w dwojaki sposób:
- przez utlenianie zwi zków zawieraj cych azot, które s obecne w paliwie (motoryzacja i ciep ownictwo komunalne i przemys owe),
- w reakcjach atmosferycznego azotu z tlenem z powietrza w temperaturach powy1ej 1300oC (du1e piece przemys owe, energetyka).
NO w atmosferze ulega licznym reakcjom – rysunek 2:
- w obecno%ci ró1nych form tlenu (O2, O3) czy rodników nadtlenkowych utlenia si. do
NO2, który jest czterokrotnie bardziej toksyczny ni1 NO’
- NO2 ulega fotochemicznemu rozk adowi daj c tlen atomowy, który w reakcji z O2
tworzy ozon. W niskich warstwach atmosfery dzia a on niszcz co na ro%liny,
- wyszczególnione tlenki mog by+ bezpo%rednio wdychane przez organizmy 1ywe. Podczas opadów s wymywane z atmosfery i w postaci kwasów wracaj na ziemi.,
Rysunek 2. Chemiczne przekszta cenia NOxw atmosferze
- z innych reakcji to NO2+ w.glowodory. Tworzy si. PAN (azotan peroksyacetylowy),
który inhibituje procesy fotosyntezy.
Zatem tlenki azotu zawarte w atmosferze s przyczyn tworzenia si. fotochemicznego smogu, kwa%nych deszczy i w konsekwencji prowadz do degradacji %rodowiska.
Emitowane tlenki azotu s bardzo szkodliwymi zwi zkami, dlatego wprowadzone s okre%lone normy dotycz ce ich emisji. Szczególne restrykcje dotycz pojazdów samochodowych – Tab.1.
Tabela 1. Europejskie normy emisji trucizn z silników Diesla [Garin F., Appl. Catal. A, 222 (2001) 183].
Normy Euro I 1993 Euro II 1996a) Euro III 2000b) Euro IV 2005c) Rodzaj emitowanych trucizn Dopuszczalne st#$enie (g/km) CO 2,72 1,0 0,64 0,50 HC - - 0,07 0,07 NOx - - 0,5-0,4 0,25-0,19 HC+NOx 0,97 0,70 0,56 0,30 CzAstki sta e 0,14 0,08 0,05-0,04 0,025-0,02
a) regulacje ECE R83.03, dyrektywa 96/69/EC
b) regulacje ECE R83.05, dyrektywa 98/69/EC; 1999/102/EC; 2001/1/EC c) regulacje ECE R83.05, dyrektywa 98/69/EC; 1999/102/EC; 2001/1/EC
W 1982 roku restrykcje obj. y silniki benzynowe, a w 1988 roku silniki Diesla. Na przestrzeni lat ulega y one drastycznym zaostrzeniom. W tabeli przedstawiono dane dotycz ce st.1e, ró1nych trucizn emitowanych z silników Diesla. W 2000 roku ilo%+ emitowanych NOx nie
mog a przekroczy+ 0,4-0,5 g/km, natomiast w 2005 roku 0,19-0,25 g/km, czyli ju1 o po ow. mniej. Wprowadzenie kolejnej normy Euro V, która wejdzie w 1ycie w roku 2009, doprowadzi do dalszych ogranicze, ilo%ci szkodliwych substancji w spalinach.
Ograniczenie emisji NOx jest bardzo wa1nym zagadnieniem. Ilo%+ wydzielanych
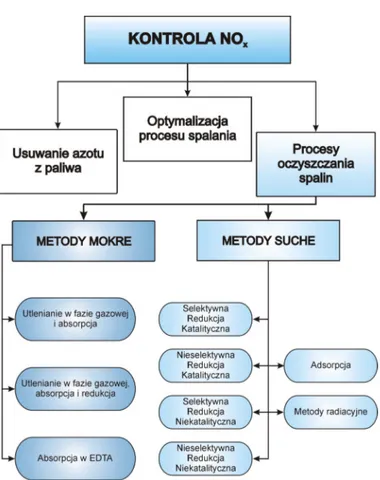
tlenków azotu mo1e by+ ograniczona poprzez zastosowanie ró1nych metod, które sprowadzaj si. do 3 zasadniczych dzia a, – rysunek 3:
Pierwsze dzia anie polega na zmniejszeniu zawarto%ci zwi zków azotu w paliwie.
Drugie, to optymalizacja procesów spalania, aby ograniczy+ tworzenie si. NOx z azotu
atmosferycznego poprzez odpowiedni konstrukcj. palenisk i palników, zachowanie optymalnego stosunku powietrze/paliwo i kontrol. temperatury w strefie spalania.
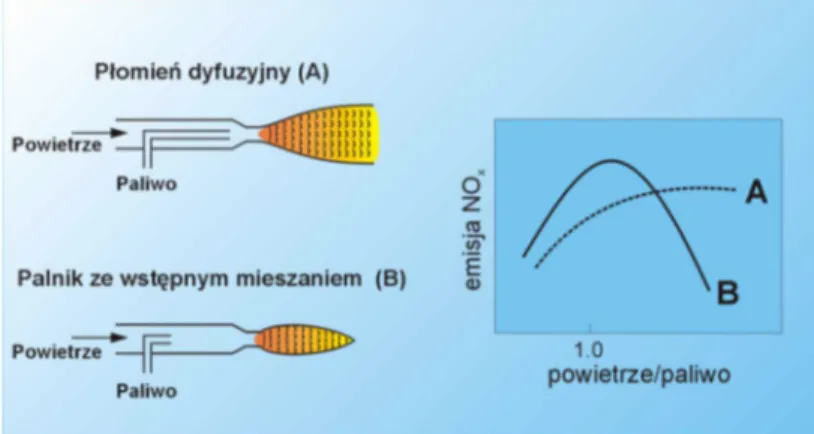
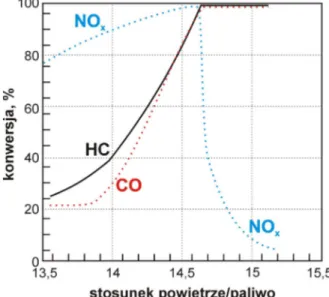
Dla podkre%lenia znaczenia kontroli procesu spalania na rys. 4. przedstawiono wykres obrazuj cy emisj. NOx w zale1no%ci od rodzaju u1ytego palnika. W przypadku A – gdy
mamy do czynienia z palnikiem spalaj cym paliwo w p omieniu dyfuzyjnym (paliwo podawane bezpo%rednio w stref. spalania jak w silniku Diesla), uzyskuje si. korzystne warunki spalania przy niskich stosunkach powietrze/paliwo. Jednak w warunkach nadmiaru powietrza znacznie korzystniejsze jest stosowanie palników ze wst.pnym mieszaniem powietrza i paliwa – B (jak w silnikach benzynowych).
Natomiast ostatni kierunek usuwania NOx obejmuje procesy oczyszczania spalin, w
sk ad których wchodz metody absorpcji w roztworach lub adsorpcji na powierzchni cia sta ych, metody radiacyjne, oraz redukcja NOxdo N2katalityczna i niekatalityczna.
Rysunek 3. Techniki pozwalaj ce ograniczy+ emisj. tlenków azotu do atmosfery
Dwa pierwsze sposoby nie s drogie i pozwalaj na zmniejszenie emisji NOx w
ilo%ciach do 50%. Natomiast trzeci sposób, najbardziej kosztowny, polegaj cy na usuwaniu NOxz gazów odlotowych, umo1liwia wyeliminowanie ich nawet w 100%.
Do najpopularniejszych metod ograniczania emisji NOxnale1 technologie bazuj ce na
reakcjach katalitycznych. W niniejszym opisie zostan omówione nast.puj ce sposoby usuwania NOxze spalin:
1. Katalityczny rozk ad NO
2. Selektywna katalityczna redukcja (SCR) 3. Nieselektywna katalityczna redukcja
4. Katalityczny rozk ad NO
Z termodynamicznego punktu widzenia bezpo%redni rozk ad NO wydaje si. najprostsz i najta,sz metod usuwania NO z gazów odlotowych.
2NO N2+ O2
Tlenek azotu jest cz steczk termodynamicznie nietrwa w niskich temperaturach w porównaniu z N2 czy O2, jednak wysoka energia aktywacji NO (364 kJ/mol) wymaga u1ycia
katalizatorów.
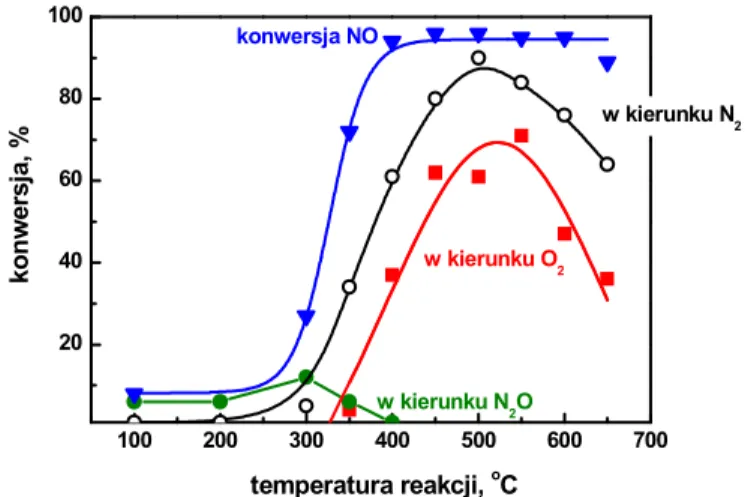
Spo%ród wielu prac po%wi.conych rozk adowi NO na metalach szlachetnych, tlenkach metali przej%ciowych i zeolitach, najbardziej obiecuj ce wyniki uzyskano dla Cu-ZSM-5 – rysunek 5. W temperaturze oko o 400oC rozk ad NO przekracza 95% jednak nie prowadzi
tylko do N2. Procesowi towarzysz reakcje uboczne - powstaj wy1sze tlenki, st d optymaln
temperatur procesu jest 500oC. Wad tych katalizatorów jest szybki spadek aktywno%ci w
Rysunek 4. Tworzenie si. tlenków azotu w zale1no%ci od rodzaju palnika i stosunku
powietrze / paliwo
5. Katalityczna redukcja tlenku azotu róCnymi czynnikami
redukujAcymi
Stosowane do redukcji tlenku azotu czynniki redukuj ce mo1na ogólnie podzieli+ na dwie grupy:
- selektywne, np. amoniak czy w.glowodory; - nieselektywne np. H2, CO.
Ró1nica mi.dzy nimi polega na tym, 1e w drugiej grupie tlen obecny w reaguj cych gazach faworyzuje reakcj. z czynnikiem redukuj cym i utlenia go. Dlatego wybieraj c katalizator do usuwania NOx nale1y pami.ta+ w jakich warunkach b.dzie on pracowa . Przyk adowo w
silnikach benzynowych, gdzie sk ad mieszaniny reakcyjnej jest bliski stechiometrycznemu stosunkowi gazów utleniaj cych do redukuj cych, faworyzowany jest proces NO+CO (ma a ilo%+ tlenu), natomiast dla silników Diesla, pracuj cych w nadmiarze tlenu, reakcja powinna by+ prowadzona z selektywnymi reduktorami.
a. Selektywna katalityczna redukcja amoniakiem
Jedn z pierwszych i obecnie najcz.%ciej stosowanych metod ograniczenia emisji tlenków azotu do atmosfery jest selektywna katalityczna redukcja amoniakiem (Selective Catalytic Reduction (SCR)). Metoda ta pierwszy raz zosta a wykorzystana w latach 70-tych w
100 200 300 400 500 600 700 20 40 60 80 100 w kierunku O2 w kierunku N2O w kierunku N2 konwersja NO ko n w er sj a, % temperatura reakcji, oC
Rysunek 5. Wp yw temperatury na aktywno%+ katalizatora Cu-ZSM-5 w reakcji rozk adu NO
Japonii. Nast.pnie wprowadzono j w USA i krajach europejskich. W 1993 roku istnia o 826 instalacji pracuj cych w oparciu o SCR.
Metoda ta polega na usuwaniu tlenków azotu z gazów odlotowych z wykorzystaniem amoniaku jako czynnika redukuj cego w obecno%ci katalizatora. Tlenki azotu ulegaj przemianie na azot i wod. wed ug nast.puj cych reakcji:
a) tlenek azotu 6 NO + 4 NH3 5 N2+ 6 H2O (1) 4 NO + 4 NH3+ O2 4 N2+ 6 H2O (2) b) dwutlenek azotu 6 NO2+ 8 NH3 7 N2+ 12 H2O (3) 2 NO2+ 8 NH3+ 4 O2 5 N2+ 12 H2O (4)
c) mieszanina stechiometryczna tlenku i dwutlenku azotu
NO2+ NO + 2 NH3 2 N2+ 3 H2O (5)
Reakcja (5) jest najszybsz reakcj w uk adzie NOx-O2-NH3. W procesie SCR tlenków azotu
w gazie spalinowym najwa1niejsz rol. odgrywa reakcja (2), poniewa1 w spalinach jest obecny tlen, a tlenek azotu stanowi zasadniczy udzia wszystkich NOx. Konieczna do zaj%cia
procesu ilo%+ amoniaku jest zbli1ona do stechiometrycznej. Ilo%+ ta powinna zapewni+ wymagan skuteczno%+ redukcji tlenków azotu przy jednoczesnym minimalnym st.1eniu nieprzereagowanego amoniaku na wylocie z reaktora. W razie niedostatecznego stopnia przereagowanego NH3 w temperaturze poni1ej 200oC w uk adzie mog powstawa+,
stwarzaj ce zagro1enie azotan amonowy (V) i azotan amonowy (III) w my%l reakcji: 2 NO2+ 2 NH3+ H2O NH4NO2+ NH4NO3 (6)
Rozk ad tych azotanów przebiegaj c wybuchowo i mo1e spowodowa+ zniszczenie instalacji oczyszczaj cej.
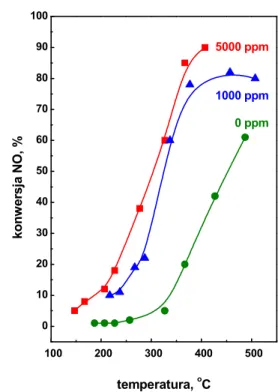
Dodatek do mieszaniny reakcyjnej tlenu w znaczny sposób podwy1sza stopie, przereagowania NO. Na rysunku 6 przedstawiono wp yw temperatury i zawarto%ci O2 na
przebieg reakcji NO+NH3 na katalizatorze V2O5/TiO2. Wraz ze wzrostem temperatury
Wydajno%+ przemiany tlenków azotu na oboj.tne sk adniki (azot i wod.) zale1y równie1 od st.1enia tlenków azotu w gazach spalinowych oraz stosunku molowego amoniak/tlenki azotu. Istotny wp yw na skuteczno%+ redukcji tlenków azotu ma te1 rodzaj i kszta t katalizatora, jego aktywno%+, selektywno%+, 1ywotno%+ i obci 1enie. Obecno%+ innych zanieczyszcze, w spalinach jak np. SO2 determinuje warunki
eksploatacyjne procesu.
Opracowano kilka technologii oczyszczania gazów z zastosowaniem selektywnej katalitycznej redukcji NOx
amoniakiem. W procesie firmy BASF AG katalizatorem jest V2O5 osadzony na Al2O3.
Warunkiem w a%ciwego przebiegu procesu jest odpowiednia (wi.ksza ni1 1% obj.) zawarto%+ O2 w gazach odlotowych z
wytwórni kwasu azotowego. Technologi. firmy Dider Engineering GmbH (Essen)
mo1na stosowa+ w fabrykach kwasu adypinowego i w elektrociep owniach. Katalizatorem jest Cr2O3 Instalacja pracuje w temperaturze ok. 280oC, katalizator jest umieszczony w
trzech warstwach, a amoniak mo1e by+ doprowadzony do ka1dej z nich. Na potrzeby przemys u krajowego opracowano technologi., w której katalizatorem jest hopkalit, pracuj cy skutecznie w temperaturach 180-250oC, pod ci%nieniem 0-700 kPa i przy du1ych
obci 1eniach gazem.
Selektywna katalityczna redukcja amoniakiem, jakkolwiek jest metod najcz.%ciej stosowan w skali przemys owej to ma jednak szereg wad:
- ze wzgl.du na rozmiary instalacji ma ograniczone zastosowanie – tylko do stacjonarnych Vróde emisji,
- mo1e by+ przyczyn dodatkowych zanieczyszcze, poprzez emisj. nieprzereagowanego NH3 lub jego wycieku czy emisj. powsta ego w wysokich
temperaturach N2O,
- nie bez znaczenia pozostaj równie1 wysokie koszty instalacji SCR.
100 200 300 400 500 0 10 20 30 40 50 60 70 80 90 100 0 ppm 1000 ppm 5000 ppm ko n w er sj a N O ,% temperatura, oC
Rysunek 6. Zale1no%+ konwersji NO w reakcji
NO+NH3od temperatury przy ró1nych
st.1eniach O2na katalizatorach V2O5/TiO2. Warunki reakcji: NO=0,5%; NH3=0,5%, szybko%C przep ywu gazów 100ml/min
[Went G.T., Leu L., Rosin R.R., Bell A.T, J. Catal.,
Dlatego wa1nym problemem staje si. znalezienie innych, równie efektywnych metod redukcji NO do N2. Bardzo obiecuj cymi metodami wydaj si. te, które zast.puj amoniak
innymi czynnikami redukuj cymi np. tlenkiem w.gla czy w.glowodorami. Ich zalet jest to, 1e CO i w.glowodory s zwykle obecne w gazach spalinowych, co zmniejsza doz. czynnika redukuj cego wymaganego do przebiegu procesu.
b. Redukcja NO w#glowodorami
Katalityczna redukcja tlenków azotu przy pomocy w.glowodorów (HC) jest równie1 atrakcyjn metod ograniczenia emisji NOx do atmosfery. Jak podkre%lono wcze%niej,
w.glowodory s zwykle obecne w gazach spalinowych, co zmniejsza ich ilo%+ wymagan do przebiegu procesu. Dodatkow korzy%ci jest eliminacja z gazów odlotowych w.glowodorów, które z tlenkami azotu tworz „smog fotochemiczny”. Pionierem bada, procesu redukcji NO w.glowodorami by M. Iwamoto. Zauwa1y on, 1e NO mo1e by+ redukowany na katalizatorze Cu-ZSM-5 przez rozmaite w.glowodory w obecno%ci tlenu. Autor ten sklasyfikowa w.glowodory na:
selektywnie dzia aj ce np.: C2H4, C3H6i C3H8które preferowa y reakcj. z NO
nieselektywne np. CH4, C2H6które w reakcji NO + w.glowodór + O2ulega y spalaniu.
Katalizatorami procesu redukcji NO w.glowodorami mog by+ uk ady zeolitowe, metaliczne i tlenkowe.
Katalizatory zeolitowe
Najcz.%ciej badanym w reakcji redukcji NO w.glowodorami jest zeolit ZSM-5. Dla preparatów typu M-ZSM-5 (M oznacza rodzaj wprowadzonego kationu) szereg aktywno%ci w reakcji NO+C2H4+O2przedstawia si. nast.puj co:
Ag=Co=Zn=Cu>H=Ni>Pt>Mn>Fe=Ca=La=Pd>Cr>Na
Z przedstawionych danych wynika, 1e jony Cu i Co s jednymi z najbardziej aktywnych, dlatego katalizatorami z tymi kationami badacze po%wi.cili najwi.cej uwagi. Jednak aktywno%+ tych wszystkich uk adów zeolitowych wyraVnie spada w obecno%ci %ladów pary wodnej czy te1 SO2. Okaza o si., 1e wprowadzenie ró1nych dodatkowych kationów np. Ba,
Katalizatory metaliczne
Zalet katalizatorów metalicznych w reakcji redukcji NO w.glowodorami w stosunku do katalizatorów zeolitowych jest odporno%+ na dzia anie pary wodnej. Posiadaj one równie1 wad.: podczas konwersji NO->N2 równolegle pojawia si. N2O (nawet 65% NO mo1e by+
konwertowane do N2O).
Spo%ród katalizatorów Pt, Ru, Rh osadzonych na -Al2O3 najwy1sz konwersj. NO
uzyskano dla Pt. Je%li chodzi o inne katalizatory metaliczne, to sporo uwagi po%wi.cono uk adom zawieraj cym Ag i Au osadzone na no%niku. I tak np. w przypadku Au osadzonego na ró1nych tlenkach stwierdzono wysok jego aktywno%+ w procesie NO+C3H6+O2. Hamada
podaje aktywno%+ ró1nych metali osadzonych na Al2O3 w reakcji NO+propan. Aktywno%ci
tych katalizatorów mo1na uszeregowa+ w nast.puj cy sposób: Co>Fe>Ni>Pt>Cu>Mn
Interesuj c grup. katalizatorów metalicznych stanowi wielosk adnikowe uk ady zawieraj ce metal szlachetny i tlenki alkaliczne osadzone na Al2O3. Okre%la si. je jako
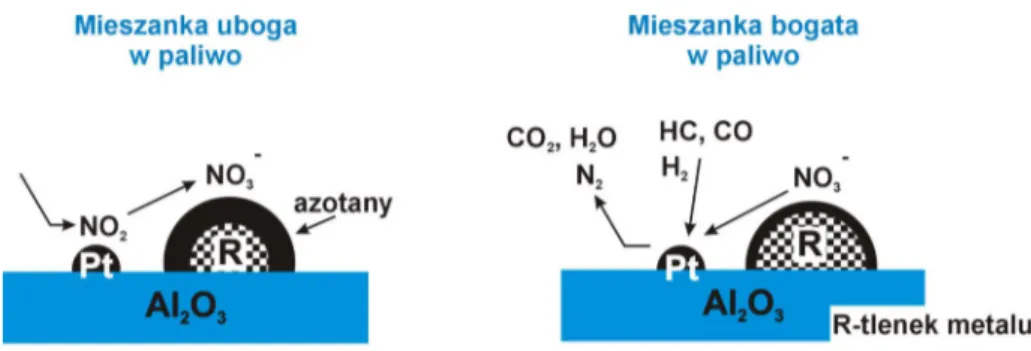
katalizatory poch aniaj co-redukuj ce (storage – reduction) W warunkach utleniaj cych (learn burn) przy du1ej zawarto%ci tlenu w gazach spalinowych i niedoborze czynników redukuj cych (mieszanka uboga w paliwo), nast.puje utlenianie tlenków azotu na metalu i ich poch anianie na zasadowym sk adniku katalizatora. Nast.pnie, gdy warunki pracy zmieniaj si. na stechiometryczne lub redukuj ce, to jest przy nadmiarze czynnika redukuj cego (wprowadzenie CO, H2, HC) (mieszanka bogata w paliwo), zaadsorbowane na
powierzchni zwi zki azotu ulegaj redukcji i usuwane zostaj z powierzchni w postaci N2,
CO2i H2O – rysunek 7.
Rysunek 7. Mechanizm dzia ania katalizatora poch aniaj co–redukuj cego [Fritz A., Pitchon V., Appl. Catal. B, 13 (1997) 1].
Katalizatory tlenkowe
Ostatnio badacze po%wi.caj coraz wi.cej uwagi tlenkom metali jako katalizatorom redukcji NO w.glowodorami. Mo1na je podzieli+ na trzy zasadnicze grupy:
a) pojedyncze tlenki metali;
b) tlenki metali osadzone na no%nikach tlenkowych;
c) tlenki metali typu perowskitu, tlenki mieszane typu spineli.
W reakcji NO+C3H8+O2 zosta y przetestowane ró1ne tlenki metali. Uzyskane rezultaty
podano w tabeli 2.
Tabela 2. Aktywno%+ katalityczna pojedynczych tlenków metali w redukcji NO propanem
(NO=1000ppm., C3H8=330ppm, O2=10% obj.)
[Hamada H., Catal. Today, 22 (1994) 21].
konwersja NO do N2(%) (konwersja C3H8do COx(%)) katalizator 200oC 300oC 400oC 500oC 600oC Cr2O3 1 (49) 21 (100) 1 (100) 0 (100) Fe2O3 0 (0) 1 (60) 0 (100) 1 (100) Co3O4 1 (72) 1 (99) 1 (100) CuO 1 (1) 1 (64) 1 (95) V2O5 0 (25) 0 (70) 0 (87) Bi2O3 0 (1) 0 (9) Al2O3 2 (1) 13 (18) 38 (93) 21 (100) ZrO2 0 (1) 10 (33) 17 (97) TiO2 1 (0) 4 (10) 8 (57) ZbO 2 (12) 4 (69) 2 (96) Y2O3 0 (1) 10 (61) 10 (94) 5 (96) La2O3 1 (8) 7 (89) 4 (100) MgO 2 (6) 8 (84) 13 (100) CaO 1 (2) 7 (27) 7 (100)
Najaktywniejszym okaza si. tlenek glinu. Tlenki metali takie jak Cr2O3, Fe2O3czy Co3O4nie
w.glowodorów. Badacze doszli do wniosku, 1e aktywnymi katalizatorami reakcji NO+HC s tlenki o kwasowych b dV zasadowych w a%ciwo%ciach powierzchni.
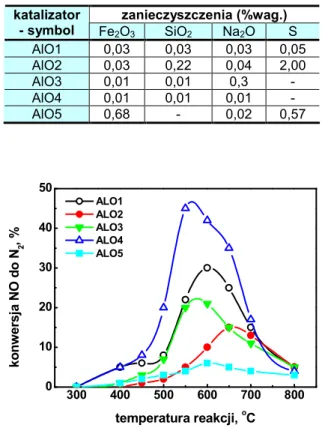
Okazaki i wspó . [Okazaki N., Shiina Y., Itoh H., Tada A., Iwamoto M., Catal. Lett., 49 (1997) 169] badaj c preparaty tlenku glinu zawieraj ce ró1ne zanieczyszczenia stwierdzili, 1e domieszki maj zasadniczy wp yw na aktywno%+ w reakcji NO+C2H4+O2. W
tabeli 3. przedstawiono ilo%+ i rodzaj zanieczyszcze, zawartych w poszczególnych katalizatorach, a na rys. 8. podano aktywno%ci tych preparatów w zale1no%ci od temperatury reakcji.
Z przedstawionych danych wynika, 1e wprowadzenie ró1nych domieszek, szczególnie Fe2O3 i tlenku siarki
powoduje obni1enie aktywno%ci tlenku glinu. W procesie redukcji tlenku azotu w obecno%ci Al2O3 stosuje si. ró1ne w.glowodory.
Bardzo dobrym okaza si. CH3OH. Porównuj c efektywno%+ redukcji NO w obecno%ci
metanolu i propanu stwierdzono, 1e u1ycie tego pierwszego pozwoli o obni1y+ temperatur. maksymalnej aktywno%ci z 550oC do ~320oC. Obecno%+ SO
2 jak i H2O w mieszaninie
reakcyjnej powodowa a spadek aktywno%ci katalizatorów monotlenkowych w reakcji redukcji NO w.glowodorami.
Tlenki metali osadzone na no nikach
W reakcji NO+HC+O2 testowano równie1 katalizatory zawieraj ce tlenki metali
osadzone na no%nikach tlenkowych czy fluorkowych. Na uwag. zas uguj badania uk adu V2O5/TiO2którego maksymalna konwersja NO do N2wynosi a ~90%. Interesuj cy okaza si.
te1 katalizator La2O3/Al2O3promotorowany strontem. W a%ciwo%ci katalityczne tych uk adów
by y znacznie lepsze ni1 Cu-ZSM-5.
Tabela 3. Sk ad zanieczyszcze, zawartych w Al2O3
[Okazaki N., Shiina Y., Itoh H., Tada A., Iwamoto M., Catal. Lett., 49 (1997) 169]. zanieczyszczenia (%wag.) katalizator - symbol Fe2O3 SiO2 Na2O S AlO1 0,03 0,03 0,03 0,05 AlO2 0,03 0,22 0,04 2,00 AlO3 0,01 0,01 0,3 - AlO4 0,01 0,01 0,01 - AlO5 0,68 - 0,02 0,57 300 400 500 600 700 800 0 10 20 30 40 50 ko n w er sj a N O d o N2 ,% temperatura reakcji, oC ALO1 ALO2 ALO3 ALO4 ALO5
Rysunek 8. Aktywno%+ ró1nych próbek Al2O3 w
reakcji NO+C2H4+O2zale1no%ci od temperatury.
[Okazaki N., Shiina Y., Itoh H., Tada A., Iwamoto M.,
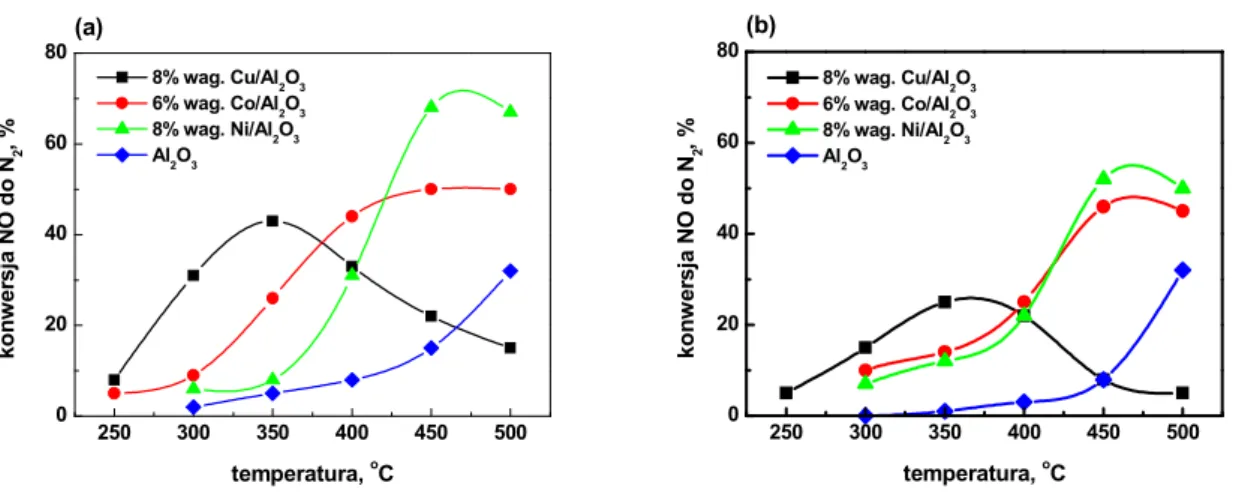
Porównano aktywno%ci uk adów Cu/Al2O3, Ni/Al2O3, Co/Al2O3w reakcji NO+C3H6+O2z
aktywno%ci samego no%nika – rys. 9. Katalizator Cu/Al2O3 wykaza najwy1sz aktywno%+
ju1 w temperaturze 350oC. Dalsze podwy1szanie temperatury powodowa o spadek konwersji
NO. Efekt ten t umaczy si. brakiem reduktora, gdy1 w temperaturze powy1ej 350oC propen
ulega ca kowitemu przereagowaniu. Natomiast katalizatory niklowy i kobaltowy dawa y wysokie przereagowanie NO do N2dopiero w temperaturze ~450oC. Jednocze%nie notowano
niski stopie, przereagowania propenu.
Dodatek do gazów reakcyjnych pary wodnej powodowa spadek aktywno%ci, co wyt umaczono blokowaniem miejsc aktywnych
przez ten reagent.
Tlenek miedzi nanoszono równie1 na inne powszechnie stosowane no%niki tlenkowe takie jak ZrO2, TiO2, SiO2, czy podwójne tlenki SiO2
-ZrO2, SiO2-ZrO2, SiO2-Al2O3czy Al2O3-Zr3(PO4)2.
U1ycie TiO2 i ZrO2 jako no%ników pozwala o na
uzyskanie katalizatorów aktywnych w usuwaniu NO z mieszaniny gazowej zawieraj cej SO2.
Tlenek siarki(IV) powodowa wzrost kwasowo%ci no%nika, a tym samym zwi.kszenie aktywno%ci uk adu.
Interesuj ce wyniki w reakcji redukcji NO propenem dla faz tlenkowych naniesionych na
250 300 350 400 450 500 0 20 40 60 80(a) ko n w er sj a N O d o N2 ,% temperatura, oC 8% wag. Cu/Al2O3 6% wag. Co/Al2O3 8% wag. Ni/Al2O3 Al2O3 250 300 350 400 450 500 0 20 40 60 80(b) 8% wag. Cu/Al2O3 6% wag. Co/Al2O3 8% wag. Ni/Al2O3 Al2O3 ko n w er sj a N O d o N2 ,% temperatura, oC Rysunek 9. Konwersja NO do N2w zale1no%ci od temperatury.
(a) NO=1000ppm, C3H6=2000ppm, O2=6,7% obj.,
(b) NO=1000ppm, C3H6=2000ppm, O2=6,7% obj., H2O=10% obj.
[Shimizu K., Maeshima H., Satsuma A., Hattori T., Appl. Catal. B, 18 (1998) 163]
0.0 0.5 1.0 1.5 2.0 0 10 20 30 40 50 60 70 80 90 100 ko n w er sj a N O d o N2 ,% czas, h MnO/MgF2 MoO3/MgF2 CuO/MgF2 CuO-MoO3/MgF2 CuO-MnO/MgF2
Rysunek 10. Aktywno%+ katalityczna
no%nik nietlenkowy, a mianowicie MgF2, uzyskano w grupie badawczej M. Wojciechowskiej –
rys. 10. Tlenki miedzi by y najaktywniejsze spo%ród badanych tlenków metali przej%ciowych, a uk ady podwójne Cu-Mn charakteryzowa y si. prawie 100% przemian NO do N2.
Tlenki typu perowskitu i tlenki mieszane typu spinelu
Tlenki typu perowskitu s efektywnymi katalizatorami redukcji NO wodorem. Sprawdzono równie1 ich aktywno%+ w reakcji redukcji tlenku azotu w.glowodorami. Perowskity zawieraj ce kobalt, miedV, lub 1elazo by y aktywne w reakcji rozk adu NO, natomiast nie wykazywa y aktywno%ci w reakcji NO+C3H6+O2. Zachodzi a tutaj
konkurencyjna, w stosunku do redukcji NO, reakcja spalania w.glowodoru. Jednak dla niektórych tlenków typu perowskitu jak La0,8Sr0,2AlO3, LaAl0,9Mg0,1O3, LaAlO3 stwierdzono
konwersj. NO do N2na poziomie 14 21%.
Aktywnymi w reakcji redukcji NO okaza y si. katalizatory otrzymane metod zol-1el. Wysoka aktywno%+ uk adu Ga2O3-Al2O3(konwersja NO do N2~100%) otrzymanego metod
zol-1el w porównaniu z pojedynczymi tlenkami glinu i galu oraz tlenkiem galu naniesionym na tlenek glinu wynika a z bardzo dobrze rozwini.tej powierzchni w a%ciwej oraz obecno%ci na powierzchni struktur typu perowskitu [GaxAl(1-x)]2O3 (x<1). Obecno%+ SO2 w gazach
reakcyjnych powodowa a spadek aktywno%ci katalizatora o zaledwie 10%.
Porównanie aktywno%ci CuAl2O4, ZnAl2O4 i MgAl2O4 w reakcji NO+C3H6/C3H8
wykaza o, 1e najlepsze stopnie przereagowania NO do N2 uzyskano dla ZnAl2O4, jednak
dopiero w temperaturze 500oC. Spinel miedziowy aktywny by ju1 w 350oC, a MgAl 2O4
maksimum aktywno%ci (~25%) przejawia w temperaturze 500oC. W produktach reakcji nie
notowano niepo1 danego N2O.
Podsumowuj c, tlenki metali przej%ciowych jako stosunkowo niedroga faza aktywna mog sta+ si. w przysz o%ci doskona ym materia em katalitycznym. Wydaje si., 1e szczególn perspektyw. maj tlenki miedzi, lantanu, chromu, podwójne uk ady tlenkowe oraz katalizatory uzyskane przez wprowadzenie niewielkich ilo%ci domieszek do fazy aktywnej (np. tlenków ceru czy manganu). Zadaniem tych ostatnich jest ustabilizowanie aktywno%ci i selektywno%ci katalizatora na wysokim poziomie.
Wykonanie /wiczenia
Celem +wiczenia jest okre%lenie aktywno%ci próbki cia a sta ego w reakcji redukcji tlenku azotu czynnikiem redukuj cym, jakim jest propen. Reakcja ta b.dzie prowadzona w sposób ci g y, w uk adzie z o1onym z mikroreaktora sprz.1onego z chromatografem gazowym. Schemat stosowanego uk adu przedstawiono poni1ej.
Schemat aparatury do redukcji tlenku azotu propenem:
.
Próbk. katalizatora (0.1g) uprzednio zaktywowanego w 400°C nale1y podpra1y+ przez 1h w temp. 300°C celem usuni.cia wilgoci.
Nast.pnie nale1y otworzy+ butl. z helem, otworzy+ kran i nastawi+ programator temperatury na po1 dan temperatur. reakcji. W tych warunkach na przep ywie helu nale1y kondycjonowa+ katalizator przez okres 45-60min.
Po up ywie tego czasu nale1y zakr.ci+ butl. z helem, zamkn + kran, nast.pnie otworzy+ butle z C3H6, NO i O2 powoli reguluj c przep yw zaworami Zimmermana tak, aby wskazania
przep ywomierzy wykazywa y po1 dane szybko%ci przep ywu. Czynno%ci te nale1y wykona+ przepuszczaj c gazy drog „poza reaktorem”.
Po ustaleniu sk adu mieszanki NO+C3H6 przekr.camy kran trójdro1ny kieruj c gazy na
reaktor. Pod wymra1alnik podstawiamy termos z ciek ym azotem. Pierwsz analiz. wykonujemy po 15 minutach od rozpocz.cia reakcji.
Po 2 minutach od wykonania nastrzyku odstawiamy ciek y azot a pod wymra1alnik nale1y podstawi+ zimn wod.. Gdy na rejestratorze chromatografu sko,czy si. zapis piku pochodz cego od NO (czas retencji ~5 minut) pod wymra1acz nale1y podstawi+ ciek y azot zamiast wody, a p.tl. nastrzykow przestawi+ w pozycj. “spulen”. Impulsy gazów poreakcyjnych na chromatograf nale1y wprowadza+ co 15min przez okres 2 godz. powtarzaj c opisan wy1ej procedur..
Opracowanie wyników
Opracowuj c wyniki nale1y obliczy+ % przemiany (konwersji) tlenku azotu do N2. Z wydruku
integratora odczytujemy powierzchni. pików NO i N2 i wpisujemy do tabeli. Wykonujemy
obliczenia konwersji oraz wykres zale1no%ci konwersji NO (o% Y) od czasu reakcji (o% X).
A B C D E F Czas (min) powierzchnia piku NO powierzchnia piku N2 2×C B+D konwersja %
D
E
×
100%
2
Zastosowanie nowych materia ów jako
katalizatorów do usuwania NO przy pomocy
tlenku w-gla
Literatura uzupe niaj ca:
Peter O’Neill, „Chemia %rodowiska” Wydawnictwo Naukowe PWN, Warszawa-Wroc aw 1998, str. 117-138.
Maria Zió ek, Izabela Nowak, „Kataliza Heterogeniczna – wybrane zagadnienia”, Wydawnictwo Naukowe UAM, Pozna4 1999, str. 118-122.
1. Wst-p
Powietrze jest jednym z najwa1niejszych elementów naszego %rodowiska; stanowi czynnik niezb.dny do 1ycia dla wi.kszo%ci znanych organizmów. Bez po1ywienia cz owiek wytrzymuje kilkana%cie dni, bez wody tylko kilka dni, a bez powietrza %mier+ nast.puje ju1 po kilku minutach. Zanieczyszczenia obecne w powietrzu wp ywaj negatywnie na rozwój i 1ycie ro%lin, zwierz t i ludzi, zagra1aj globalnemu systemowi klimatycznemu; a niszczenie ozonu stratosferycznego jest groVne dla wszystkich organizmów 1ywych na ca ej kuli ziemskiej. Nale1y zaznaczy+, 1e z punktu widzenia rolnictwa zanieczyszczenie powietrza jest o tyle groVne, 1e przez ten element %rodowiska ska1enia przenoszone zostaj na inne sk adniki ekosystemu tj. gleb., wody powierzchniowe, ro%liny.
Tlenki azotu nale1 do najbardziej uci 1liwych zanieczyszcze, gazowych atmosfery. Mog one wyst.powa+ w rozmaitych formach, takich jak: N2O, NO, N2O3, NO2, N2O4, N2O5,
NO3, N2O6, jednak w powietrzu atmosferycznym najwi.cej jest NO i NO2. Ich sum. przyj.to
okre%la+ jako NOx. G ówne Vród o emisji NOx 90-95% stanowi NO. Pochodzi on g ównie ze
spalania paliw naturalnych takich jak w.giel i paliw w pojazdach samochodowych.
Tlenek azotu tworzy si. zasadniczo w dwojaki sposób:
- z jednej strony przez utlenianie zwi zków zawieraj cych azot, które s obecne w paliwie
- a z drugiej w reakcjach atmosferycznego azotu z tlenem obecnym w powietrzu w temperaturach powy1ej 1300oC.
Utworzony tlenek azotu w atmosferze ulega licznym reakcjom chemicznym prowadz cym do powstania ró1norakich, cz.sto bardziej szkodliwych zwi zków:
- w obecno%ci O2, O3czy rodników nadtlenkowych utlenia si. do NO2
- NO2 ulega fotochemicznemu rozk adowi daj c tlen atomowy, który w reakcji z O2
tworzy ozon. W niskich warstwach atmosfery dzia a on niszcz co na ro%liny
- wyszczególnione tlenki mog by+ bezpo%rednio wdychane przez organizmy 1ywe. Podczas opadów s wymywane z atmosfery i w postaci kwasów wracaj na ziemi.. - z innych reakcji to NO2+ w.glowodory. Tworzy si. PAN (azotan peroksyacetylowy),
który inhibituje procesy fotosyntezy.
Emitowane tlenki azotu s bardzo szkodliwymi zwi zkami, dlatego stosuje si. ró1ne metody prowadz ce do ich ograniczenia (patrz +wiczenie 1). Najefektowniejszymi z tych metod s dzia ania maj ce na celu oczyszczanie spalin, a w%ród nich metody katalityczne. Procesy katalityczne s najbardziej kosztowne, ale pozwalaj na wyeliminowanie NOx z
gazów odlotowych nawet w 100%. Jednym z takich katalitycznych sposobów jest redukcja tlenków azotu ró1nymi czynnikami redukuj cymi. Mo1na je podzieli+ na selektywne jak amoniak, w.glowodory i nieselektywne, do których nale1 H2 czy CO. Zalet tlenku w.gla
jest jego obecno%+ w gazach spalinowych, co zmniejsza doz. czynnika redukuj cego wymaganego do przebiegu procesu.
2. Katalityczna redukcja tlenku azotu tlenkiem w-gla
Tlenek w.gla jest jednym z najbardziej efektywnych reduktorów tlenków azotu. Jednak w obecno%ci tlenu proces ten jest bardzo trudny do przeprowadzenia z uwagi na reakcj. CO z tlenem. Najwy1sze stopnie przemiany NO do N2 uzyskuje si. przy zawarto%ci O2 poni1ej
1% obj. i temperaturze reakcji 150-350oC.
Katalizatory metaliczne
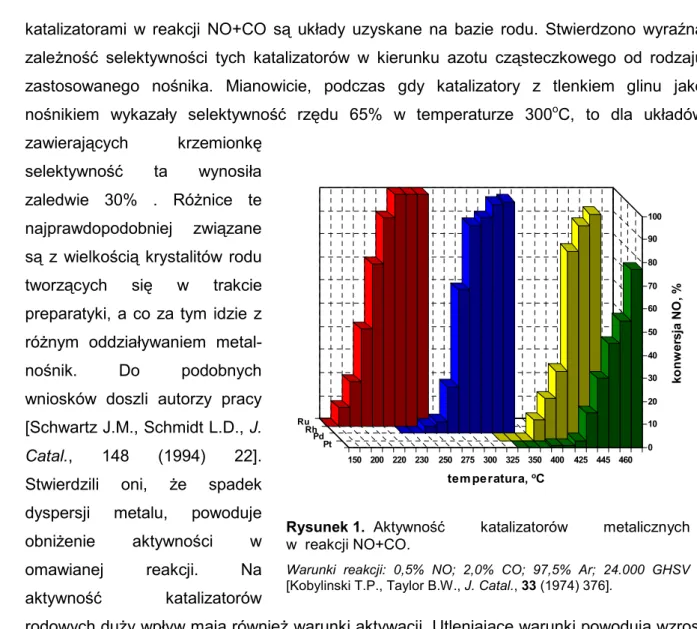
Ju1 w latach 70-tych badacze wskazali, 1e katalizatory zawieraj ce ruten i rod odznaczaj si. dobrymi w a%ciwo%ciami w reakcji redukcji NO tlenkiem w.gla. Autorzy tych prac ustalili nast.puj cy szereg aktywno%ci: Ru>Rh>Pd>Pt – rys. 1. Uk ady rutenowe s najbardziej aktywne, jednak ich aktywno%+ jest niestabilna. W trakcie reakcji tworz si. lotne
katalizatorami w reakcji NO+CO s uk ady uzyskane na bazie rodu. Stwierdzono wyraVn zale1no%+ selektywno%ci tych katalizatorów w kierunku azotu cz steczkowego od rodzaju zastosowanego no%nika. Mianowicie, podczas gdy katalizatory z tlenkiem glinu jako no%nikiem wykaza y selektywno%+ rz.du 65% w temperaturze 300oC, to dla uk adów
zawieraj cych krzemionk. selektywno%+ ta wynosi a zaledwie 30% . Ró1nice te najprawdopodobniej zwi zane s z wielko%ci krystalitów rodu tworz cych si. w trakcie preparatyki, a co za tym idzie z ró1nym oddzia ywaniem metal-no%nik. Do podobnych wniosków doszli autorzy pracy [Schwartz J.M., Schmidt L.D., J. Catal., 148 (1994) 22]. Stwierdzili oni, 1e spadek dyspersji metalu, powoduje obni1enie aktywno%ci w omawianej reakcji. Na aktywno%+ katalizatorów
rodowych du1y wp yw maj równie1 warunki aktywacji. Utleniaj ce warunki powoduj wzrost aktywno%ci i stabilizacj. uk adu katalitycznego.
Inn grup. katalizatorów metalicznych stanowi uk ady podwójne Pt-Rh, Pd-Rh, Pd-Cr czy Pd-Ag na no%nikach tlenkowych. Niejednokrotnie wykazuj one wy1sz aktywno%+ i selektywno%+ oraz stabilno%+ czy odporno%+ na zatrucia ni1 uk ady zawieraj ce tylko jeden ze sk adników aktywnych. Uk ady takie wykorzystywane s jako katalizatory trójfunkcyjne w pojazdach samochodowych. Istotn rol. odgrywa w nich tworzenie si. powierzchniowych stopów.
Katalizatory trójfunkcyjne
Katalizatory trójfunkcyjne (THREE-WAY CATALYST – TWC) s u1 do eliminacji trzech sk adników obecnych w gazach odlotowych tj.: NOx, CO i w.glowodorów. Reakcje
towarzysz ce temu procesowi mo1na opisa+ nast.puj co:
0 10 20 30 40 50 60 70 80 90 100 ko n w er sj a N O ,% Ru Rh Pd Pt 150 200 220 230 250 275 300 325 350 400 425 445 460 tem peratura, oC
Rysunek 1. Aktywno%+ katalizatorów metalicznych
w reakcji NO+CO.
Warunki reakcji: 0,5% NO; 2,0% CO; 97,5% Ar; 24.000 GHSV
2NO + 2CO N2+ 2CO2
CO + 1/2 O2 CO2
CxHy+ (x + y/4)O2 xCO2+ y/2H2O
Stopie, przereagowania poszczególnych sk adników zale1y w du1ej mierze od sk adu spalin, a co za tym idzie od stosunku powietrze/paliwo – rys. 2. Tlenek w.gla i w.glowodory przereagowuj ca kowicie w warunkach utleniaj cych, podczas gdy NOx w warunkach
redukuj cych. Wynika st d, 1e aby katalizator pracowa w sposób efektywny musi by+ zapewniony odpowiedni stosunek powietrze/paliwo (lub wspó czynnik , definiowany jako stosunek powietrza dost.pnego do powietrza potrzebnego do ca kowitego spalenia paliwa, najlepiej, gdy
=1).
Katalizatory trójfunkcyjne s uk adami stanowi cymi kombinacj. metali szlachetnych takich jak platyna i rod osadzonych na monolicie kordierytu – 2MgO, 5SiO2 i 2Al2O3 stabilizowanym
przez zasadowe tlenki metali tj. baru, lantanu czy sodu. O wyborze metali szlachetnych jako fazy aktywnej
zadecydowa y dwa czynniki: wysoka aktywno%+ w procesie redukcji NOx po bardzo krótkim
czasie kontaktu z katalizatorem, oraz odporno%+ na trucizny zawarte w gazach spalinowych, szczególnie na SO2. Do katalizatorów dodaje si. równie1 cer jako promotor fazy aktywnej.
Zapewnia on dobr dyspersj. metalu, wzrost stabilno%ci termicznej Al2O3, przyspiesza
usuwanie CO w warunkach utleniaj cych oraz u atwia adsorpcj. i desorpcj. tlenu w zale1no%ci od warunków pracy katalizatora. Podobn rol. jak cer spe niaj tlenki manganu wprowadzone do katalizatora TWC.
Dodatek dwóch tlenków CeO2+ ZrO2 powoduje stabilizacj. oddzia ywa, tego pierwszego z
metalem szlachetnym. Dodatkowo zapewnia wy1sz odporno%+ termiczn ca ego uk adu. Pierwsze katalizatory pe ni ce rol. utleniaczy tlenku w.gla(II) i w.glowodorów wprowadzone zosta y w USA w roku 1975. Natomiast w Europie tego typu katalizatory wykorzystano dopiero w latach 80-tych. Obecnie dla silników benzynowych stosuje si. katalizatory o potrójnym dzia aniu: usuwaj CO, tlenki azotu i niespalone w.glowodory.
Rysunek 2. Zale1no%+ konwersji NOx, CO i HC
od stosunku powietrze/paliwo.
stechiometrycznym stosunku ilo%ci gazów utleniaj cych do redukuj cych, co kontroluje si. stosunkiem dop ywu powietrze/paliwo. Jednak nadal poszukuje si. nowych katalizatorów dla tych silników.
Katalizatory tlenkowe
Innymi katalizatorami aktywnymi w reakcji redukcji tlenków azotu tlenkiem w.gla s tlenki metali. Pierwsze dane o ich aktywno%ci pojawi y si. ju1 w latach 60-tych. Shelef i wspó pracownicy wykazali ró1n przydatno%+ tlenków metali osadzonych na tlenku glinu w tej reakcji - rys. 3. Najwy1sz aktywno%ci odznacza si. Fe2O3.
Tlenek chromu by mniej aktywny ani1eli NiO. W przypadku tego ostatniego azot cz steczkowy
tworzy si. w temperaturze 325oC, a 100%-ow konwersj. do N
2rejestrowano w 470oC. Dla
Cr2O3 temperatury te wynosi y odpowiednio 430oC i 500oC. Dodatek tlenu cz steczkowego
do reagentów obni1a aktywno%+ w przypadku obydwu tlenków. Spadek aktywno%ci NiO t umaczono efektem konkurencyjnego oddzia ywania pomi.dzy O2 i CO, co prowadzi o
do utleniania czynnika redukuj cego. Równie1 dodatek pary wodnej do mieszaniny reakcyjnej powodowa wyraVny spadek aktywno%ci. I tak, w obecno%ci pary wodnej dla katalizatora chromowego 90%-ow aktywno%+ osi gano dopiero w temperaturze 520oC,
podczas gdy przy jej braku ju1 w 325oC. Podobna sytuacja mia a miejsce dla uk adu
Fe2O3/Al2O3– temperatury wynosi y odpowiednio 280oC i 485oC.
Aktywnymi w reakcji redukcji NO tlenkiem w.gla okaza y si. równie1 uk ady podwójne zawieraj ce tlenki miedzi jako jeden ze sk adników aktywnych. Wykazano wy1sz aktywno%+ katalizatorów CuO/CeO2 lub CuO/CeO2- -Al2O3 ni1 samego CuO, co wyt umaczono lepsz
dyspersj CuO w uk adach podwójnych.
Inni badacze prowadzili eksperymenty dotycz ce reakcji NO+CO z udzia em tlenku miedzi i manganu osadzonych na w.glu aktywnym. Preparaty miedziowe charakteryzowa y si. wysok , blisko 100%-ow konwersj. NO do N2w temperaturze 175oC, podczas gdy uk ady z
0 100 200 300 400 500 600 0 20 40 60 80 100 Co3O4 Cu2O Fe2O3 MnO NiO Cr2O3 V2O5 ko n w er sj a N O ,% temperatura, oC
Rysunek 3. Aktywno%+ ró1nych tlenków metali w reakcji
NO+CO.
Warunki reakcji: CO~1,2%, NO~2,0%, szybko%C przep ywu 1400 ml/min
tlenkiem manganu jako faz aktywn dopiero w 300oC dawa y przereagowanie rz.du
65-70%. Wprowadzenie obu sk adników równocze%nie pozwoli o uzyska+ 100% konwersj. w temperaturze 250oC. Na powierzchni katalizatora tworzy si. spinel typu CuMn2O4
odpowiedzialny za aktywno%+. Podobne rezultaty otrzymano dla uk adu miedziowo – manganowego osadzonego na Al2O3.
Z kolei katalizatory PdO-MoO3/Al2O3 by y odporne na dzia anie tlenu, podczas gdy uk ady
pojedyncze ulega y atwo dezaktywacji w obecno%ci tego czynnika.
3. Mechanizm redukcji tlenku azotu tlenkiem w-gla
Bardzo wa1nym etapem w poszukiwaniu nowych, aktywnych katalizatorów reakcji redukcji NO tlenkiem w.gla jest poznanie mechanizmu tej reakcji. Pomimo wielu prac, jakie ukaza y si. na ten temat nie jest on do ko,ca poznany. Mechanizm reakcji zale1y od wielu czynników takich jak: rodzaj fazy aktywnej, no%nik czy sk ad mieszaniny reakcyjnej.
Reakcja redukcji NO tlenkiem w.gla sk ada si. z dwóch jednoczesnych procesów: redukcji NO do N2i utlenienia CO do CO2.Sumarycznie te dwa procesy mo1na przedstawi+
nast.puj co:
2
NO
+
2
CO
kat.N
2+
2
CO
2Jednak1e w procesie tym mo1e równie1 tworzy+ si. N2O w trakcie nieca kowitej redukcji NO:
2
NO CO
+
kat.N O CO
2+
2Mechanizmy procesów prowadz ce do tworzenia wymienionych produktów mo1na przedstawi+ w nast.puj cy sposób:
CO
S
CO
NO
S
NO
NO
S
N
O
CO
O
CO
S
NO
N
N O
N O
N
O
N O
N O
S
N
N
N
S
g g g g g g+
+
+
+
+
+
+
+
+
+
+
* * * * * * * * * * * * * * * 2 2 2 2 2 2 22
gdzie: (1) (2) (3) (4) (5) (6) (7) (8)g - reagent gazowy * - adsorbowany S - miejsce aktywne
Jak wynika z powy1szych równa,, pierwszym a zarazem koniecznym etapem redukcji tlenku azotu tlenkiem w.gla jest adsorpcja reagentów na powierzchni katalizatora (równania 1,2). W dalszych etapach reakcja mo1e zachodzi+ wed ug mechanizmu jednocz steczkowego b dV dwucz steczkowego. Wed ug mechanizmu jednocz steczkowego nast.puje reakcja zaadsorbowanej cz steczki CO z zaadsorbowanym atomem tlenu pochodz cym z dysocjacyjnej chemisorpcji tlenku azotu. W przypadku mechanizmu dwucz steczkowego reakcja przebiega pomi.dzy zaadsorbowanymi cz steczkami CO i NO.
Najwa1niejszym stadium reakcji jest dysocjacja zaadsorbowanego tlenku azotu (równanie 3). W nast.pnej kolejno%ci adsorbowany atom azotu ulega rekombinacji do N2,
cz c si. z drugim takim samym atomem (równanie 8). Z kolei zaadsorbowany atom tlenu cz c si. zaadsorbowan cz steczk CO desorbuje si. w postaci CO2(równanie 4).
Obecno%+ w produktach reakcji podtlenku azotu zwi zana jest z reakcj zaadsorbowanego atomu azotu z zaadsorbowan cz steczk NO (równanie 5). Powsta y w tym etapie adsorbowany N2O mo1e ulec rozk adowi z wytworzeniem gazowego N2, b dV
desorpcji daj c gazowy N2O (równanie 6, 7). Kierunek tej reakcji zale1y od szybko%ci tych
dwóch procesów. Je1eli szybko%+ rozk adu jest znacznie szybsza od szybko%ci desorpcji N2O, wówczas w produktach reakcji nie obserwuje si. podtlenku azotu.
O tym, czy reakcja b.dzie jednocz steczkowa czy dwucz steczkowa w du1ej mierze decydowa+ b.d w a%ciwo%ci katalizatora. Mianowicie, je1eli proces dysocjacji powierzchniowego NO* b.dzie zachodzi bardzo szybko, wówczas znacznie zmniejsza si. prawdopodobie,stwo zaj%cia reakcji dwucz steczkowej. Natomiast w przypadku, gdy dysocjacja NO* b.dzie przebiega+ stosunkowo wolno, to prawdopodobie,stwo zaj%cia reakcji z udzia em zaadsorbowanych CO i NO b.dzie bardzo du1e. St d mo1na stwierdzi+, 1e powierzchniowa dysocjacja NO* b.dzie etapem limituj cym szybko%+ przebiegu procesu. Oprócz N2, który jest g ównym i po1 danym produktem reakcji redukcji NO tlenkiem
w.gla, w procesie tym mo1e równie1 powstawa+ N2O. Tlenek azotu(I) by obserwowany w
zale1no%ci od temperatury reakcji. Takie zachowanie si. katalizatorów nasun. o przypuszczenie, 1e obydwa produkty, tj. N2O i N2mog mie+ wspólny zwi zek przej%ciowy –
N2O*. Tworzenie si. takiego kompleksu powierzchniowego, wyja%nia obecno%+ lub brak w
produktach reakcji podtlenku azotu. Zatem obecno%+ N2O w produktach reakcji jest
reakcji redukcji NO tlenkiem w.gla jest reakcja pomi.dzy zaadsorbowanymi atomami azotu i desorpcja N2.
Wykonanie /wiczenia
Celem +wiczenia jest zbadanie mo1liwo%ci wykorzystania nowego materia u jako katalizatora redukcji tlenku azotu tlenkiem w.gla. W tym celu zostanie okre%lona aktywno%+ danej próbki w tej reakcji. Proces NO+CO b.dzie prowadzony w sposób ci g y, w uk adzie z o1onym z mikroreaktora sprz.1onego z chromatografem gazowym. Schemat stosowanego uk adu przedstawiono poni1ej.
Schemat aparatury do redukcji tlenku azotu tlenkiem wMgla:
Próbk. cia a sta ego (0.1g) uprzednio zaktywowan w 400°C nale1y podpra1y+ przez 1h w temp. 300°C celem usuni.cia wilgoci.
Nast.pnie nale1y otworzy+ butl. z helem, otworzy+ kran i nastawi+ programator temperatury na po1 dan temperatur. reakcji. W tych warunkach na przep ywie helu nale1y kondycjonowa+ katalizator przez okres 45-60min.
Po up ywie tego czasu nale1y zakr.ci+ butl. z helem, zamkn + kran, nast.pnie otworzy+ butle z CO, NO i O2 powoli reguluj c przep yw zaworami Zimmermana tak, aby wskazania
przep ywomierzy wykazywa y po1 dane szybko%ci przep ywu. Czynno%ci te nale1y wykona+ przepuszczaj c gazy drog „poza reaktorem”.
Po ustaleniu sk adu mieszanki NO+CO przekr.camy kran trójdro1ny kieruj c gazy na reaktor. Pod wymra1alnik podstawiamy termos z ciek ym azotem. Pierwsz analiz. wykonujemy po 15 minutach od rozpocz.cia reakcji. Po 2 minutach od wykonania nastrzyku odstawiamy ciek y azot a pod wymra1alnik nale1y podstawi+ zimn wod.. Gdy na rejestratorze chromatografu sko,czy si. zapis piku pochodz cego od NO (czas retencji ~5 minut), pod wymra1acz nale1y podstawi+ ciek y azot zamiast wody, a p.tl. nastrzykow przestawi+ w pozycj. “spulen”. Impulsy gazów poreakcyjnych na chromatograf nale1y wprowadza+ co 15min przez okres 2 godz. powtarzaj c opisan wy1ej procedur..
Opracowanie wyników
Opracowuj c wyniki nale1y obliczy+ % przemiany (konwersji) tlenku azotu do N2. Z wydruku
integratora odczytujemy powierzchni. pików NO i N2 i wpisujemy do tabeli. Wykonujemy
obliczenia konwersji oraz wykres zale1no%ci konwersji NO (o% Y) od czasu reakcji (o% X).
A B C D E F Czas (min) powierzchnia piku NO powierzchnia piku N2 2×C B+D konwersja %
D
E
×
100%
3
Wykorzystanie cia sta ych w badaniach
mechanizmów reakcji katalitycznych.
Mechanizm redukcji NO propenem.
Literatura uzupe niaj ca:
Peter O’Neill, „Chemia %rodowiska” Wydawnictwo Naukowe PWN, Warszawa-Wroc aw 1998, str. 117-138.
Maria Zió ek, Izabela Nowak, „Kataliza Heterogeniczna – wybrane zagadnienia”, Wydawnictwo Naukowe UAM, Pozna4 1999, str. 118-122.
Wa1nym etapem bada, katalizatorów w reakcji redukcji tlenku azotu w.glowodorami jest poznanie mechanizmu, wed ug którego proces ten zachodzi. Niniejsze +wiczenie przedstawia skondensowany opis mechanizmów reakcji redukcji tlenku azotu w.glowodorami, proponowanych dla ró1nych katalizatorów. Odno%nie mechanizmu reakcji redukcji NO w.glowodorami, to proponowane s ró1ne mechanizmy w zale1no%ci od rodzaju katalizatora, warunków prowadzenia reakcji, temperatury czy zawarto%ci tlenu. Ogólnie mo1na wyró1ni+ trzy kierunki:
1. Cz steczka NO zostaje utleniona przez tlen do NO2, która nast.pnie jako silny czynnik
utleniaj cy reaguje z w.glowodorami powoduj c uwolnienie CO2i N2.
NO + O2 NO2(ads) NO2(ads) + CH4 [X] + nH2O [X] + NO N2+ CO + mH2O [X] + NO2 N2+ CO2+ mH2O [X] – produkt przej%ciowy Spis tre%ci
2. Cz steczka w.glowodoru zostaje najpierw cz.%ciowo utleniona przez tlen, a nast.pnie ma zdolno%+ do selektywnego redukowania NO do N2. Rezultatem tych oddzia ywa, jest
tworzenie ró1nych zwi zków przej%ciowych, które w wyniku rozpadu daj N2i CO2.
C3H6(ads) + 1/2O2 C3OH6(ads)
C3OH6(ads) + 8NO(ads) 4N2+ 3CO2+ 3H2O
3. Proces redukcji NO w.glowodorami zachodz cy wg mechanizmu redox obejmuje kolejno utlenianie i redukcj. powierzchni katalizatora tlenkiem azotu i w.glowodorami.
9 O-Cu2+ + C
3H6 9 Cu++ 3 CO2+ H2O
2 NO-Cu+ N
2+ 2 O-Cu2+
Generalnie mówi c, procesy redox na powierzchni cia sta ych polegaj na przej%ciu jednego elektronu z zaadsorbowanej cz steczki do centrum utleniaj cego na powierzchni (utlenianie), lub z centrum redukuj cego cia a sta ego do cz steczki zaadsorbowanej (proces redukcji). Tak wi.c aby na ciele sta ym zasz a reakcja utleniania b dV redukcji musz si. znajdowa+ na jego powierzchni miejsca zdolne odpowiednio do przyj.cia lub oddania jednego elektronu. Rol. centrów elektronoakceptorowych na powierzchni preparatów mog pe ni+ np. jony metalu zdolne do przej%cia na ni1szy stopie, utlenienia. Przyk adem takich centrów mog by+ jony Cu2+ czy te1 Cr6+. W pewnych sytuacjach centrami redukuj cymi
mog by+ równie1 centra kwasowe Lewisa (tzn. centra zdolne do przyj.cia pary elektronowej). Natomiast centrami jednoelektronodonorowymi b.d centra metaliczne zdolne do przej%cia na wy1szy stopie, utlenienia jak: Cu0, Cu1+ czy Cr3+. Ponadto rol. tego rodzaju
centrów mog równie1 spe nia+ miejsca zasadowe Lewisa (zdolne do oddania pary elektronowej).
Utlenianie tlenku azotu i redukcja NO
2w-glowodorem
Badaj c mechanizm reakcji NO+C3H6 na katalizatorach CuO/Al2O3 stwierdzono, 1e
pierwszym etapem procesu jest utlenienie Cuo/Cu+do Cu2+ za pomoc NO i O
2zawartych w
gazach reakcyjnych. Nast.pnym etapem jest adsorpcja na jonach Cu2+ tlenku azotu(IV)
powsta ego w wyniku utleniania NO w fazie gazowej. Schemat przebiegu reakcji przedstawiony zosta na rys. 1.
Obecno%ci NO2 na powierzchni katalizatorów tlenkowych wykazali Haneda i wspó .
[Haneda M., Kintaichi Y., Inaba M., Hamada H., Catal. Today, 42 (1998) 127]. Autorzy ci badaj c uk ad Ag/TiO2-ZrO2
stwierdzili, 1e powstaj cy na kwasowych miejscach no%nika NO2
jest redukowany do N2 poprzez
reakcj. z C3H6 na centrach
srebrowych. Podobnie autorzy pracy [Coronado J.M., Anderson J.A., J. Mol. Catal. A, 138 (1999) 83] badaj c uk ad Pt/BaCl2/SiO2 wykazali, 1e
pierwszym etapem reakcji jest utlenienie NO do NO2. Nast.pnie NO2
ulega reakcji z w.glowodorem.
Do interesuj cych wniosków na temat mechanizmu reakcji NO+C3H6
doszli równie1 Yokoyama i wspó . [Yokoyama C., Misono M., Catal. Lett., 29 (1994) 1] badaj c uk ad Ce-ZSM-5 promotorowany Mn2O3. U1ycie mieszaniny Ce-ZSM-5 i Mn2O3
pozwala o osi gn + znacznie wy1sze przemiany NO do N2 ni1 na samym Ce-ZSM-5.
Uzyskanie wysokiej aktywno%ci autorzy t umacz tym, 1e na tlenku manganu(III) nast.puje utlenienie NO do NO2. Istniej równie1 opinie, 1e utlenianie NO do NO2nie nast.puje w fazie
gazowej, a ma miejsce na cerowych miejscach aktywnych.
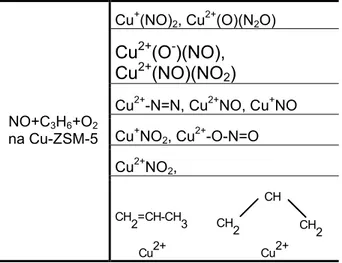
Inni autorzy badaj c uk ady Cu-ZSM-5 w reakcji NO+HC wykazali obecno%+ kompleksu Cu2+-O-N=O na powierzchni katalizatora. Stwierdzili równie1 istnienie szeregu
innych zwi zków przej%ciowych, które zestawiono w tabeli 1.
Kolejny etap reakcji stanowi utlenienie zaadsorbowanego NO2do grup azotanowych:
Tabela 1. ZwiNzki przej%ciowe powstajNce podczas
redukcji NO propenem w obecno%ci tlenu.
Cu+(NO)2, Cu2+(O)(N2O)
Cu
2+(O
-)(NO),
Cu
2+(NO)(NO
2)
Cu2+-N=N, Cu2+NO, Cu+NO Cu+NO2, Cu2+-O-N=O Cu2+NO2, NO+C3H6+O2 na Cu-ZSM-5 Cu2+ CH 2=CH-CH3 CH2 CH CH 2 Cu2+Rysunek 1. Schemat mechanizmu reakcji NO+C3H6na katalizatorze Cu/Al2O3
NO N2O3 N2O4 NO2 NO3
-Przyk adowo poni1ej podano szereg reakcji prowadz cych do utworzenia grup azotanowych na powierzchni katalizatora Cu/Al2O3. Utworzone azotany(V) i azotany(III)
reaguj z w.glowodorami, tworz c zwi zki przej%ciowe typu: –NCO, -CN, CH3COO-, R-NH2,
CH2=CH-NO2, CH3NO2, które nast.pnie ulegaj przemianie do azotu cz steczkowego, CO2
i wody.
Adsorpcja w-glowodoru
i redukcja tlenku azotu
Wielu badaczy uwa1a, 1e kluczowym etapem reakcji NO+HC jest utworzenie cz.%ciowo utlenionej formy w.glowodoru, która nast.pnie redukowana jest przez tlenek azotu. Ko,cowe produkty stanowi N2, CO2
i H2O.
Najpierw nast.puje adsorpcja w.glowodoru na powierzchni katalizatora. Do wniosku takiego doprowadzi y badania mechanizmu reakcji NO+C3H6+O2 na Cu-ZSM-5.
Propen adsorbowany w niskich temperaturach (~300oC) ulega
utlenieniu do kwasów
Rysunek 2. Schemat mechanizmu reakcji
NO+C3H6+O2na katalizatorze Cu-ZSM-5
karboksylowych. Schemat mechanizmu przedstawiono na rys. 2. Utworzenie kwasów karboksylowych mo1liwe jest dzi.ki obecno%ci na powierzchni katalizatora klasterów miedziowych zawieraj cych mostki tlenowe typu Cu-O-Cu, lub grupy hydroksylowe Cu-OH. Gdy w gazach reakcyjnych znajduje si. NO, wtedy wymienione powy1ej ugrupowania miedziowo – tlenowe mog by+ Vród em tlenu potrzebnego do utlenienia tlenku azotu do NO2. Ten ostatni du1o atwiej reaguje z zaadsorbowanymi w.glowodorami tworz c zwi zki
przej%ciowe typu R-NHOH, R-CN.
Obserwowano równie1 tworzenie si. izocyjanianów jako zwi zków przej%ciowych w reakcji NO+HC np. na powierzchni katalizatora Ag/Al2O3. Badacze sugeruj , 1e tlenek azotu
reaguje z zaadsorbowanymi grupami –NCO tworz c azot cz steczkowy.
Inni badacze rozwa1aj c mechanizm reakcji NO+HC uwa1aj , 1e w.glowodory mo1na podzieli+ na dwie zasadnicze grupy. Te, które s aktywne w reakcji z NO i takie, które nie reaguj z tlenkiem azotu. Te ostatnie staj si. aktywne dopiero po utlenieniu na powierzchni katalizatora. Produktem reakcji zaadsorbowanego w.glowodoru z NO jest cz steczkowy azot i najcz.%ciej powierzchniowe grupy –NCO, mog ce reagowa+ dalej z NO tworz c N2 i
CO2.
Rozk ad NO na powierzchni katalizatora, desorpcja N
2i utlenienie
HC przez zaadsorbowany na miejscach aktywnych tlen
Burch i Scire [Burch R., Scire S., Appl. Catal. B, 3 (1994) 295] przeprowadzili badania redukcji NO metanem b dV etanem na katalizatorach Cu-, Co-, Rh- i Pt-ZSM-5. Stwierdzili oni, 1e mechanizm reakcji jest podobny do tego, jaki ma miejsce w przypadku rozk adu NO. Tlenek azotu ulega redukcji na centrach metalicznych. Na powierzchni katalizatora pozostaje zaadsorbowany tlen, natomiast azot ulega desorpcji w postaci cz steczkowej. Zaadsorbowany tlen usuwany jest z powierzchni katalizatora przez w.glowodór. Nast.puje regeneracja miejsca aktywnego. Podobnie w przypadku uk adów platynowych badacze stwierdzili, 1e kluczowym etapem reakcji jest rozk ad NO.
Do podobnych wniosków doszli Inui i wspó . [Inui T., Iwamoto S., Kojo S., Yoshida T., Catal. Lett., (1992) 1063] badaj c redukcj. NO cetanem. Badacze obserwowali obecno%+ niewielkich ilo%ci aldehydów i ketonów na powierzchni katalizatorów zeolitowych typu Cu-Na-A. Sugerowa o to, 1e zwi zki przej%ciowe powstaj w wyniku reakcji w.glowodoru z zaadsorbowanym tlenem, podczas gdy azot ulega desorpcji. W.glowodór ulegaj c utlenieniu redukowa miejsca aktywne katalizatora powoduj c ich regeneracj..
Wykonanie /wiczenia
Badania spektroskopowe w podczerwieni wykonuje si. na spektrometrze firmy Bio-Rad, model Excalibur 3000 z transformacj Fouriera (FT-IR).
Katalizator miedziowy np. tlenek miedzi osadzony na MgF2, w postaci tabletki
(~4 mg/cm2) nale1y sprasowa+ pod ci%nieniem 20 MPa, a nast.pnie umie%ci+ w kuwecie
pró1niowej zaopatrzonej w okienka z KRS5. Próbk. nale1y zaktywowa+ w temperaturze 150°C przez 30 min, oraz odgazowywa+ przy ci%nieniu 10-4 Torr, a nast.pnie ch odzi+ do
temperatury pokojowej i zarejestrowa+ widmo podstawowe.
Nast.pnie do kuwety wprowadza si. kolejno adsorbowane gazy: NO, C3H6 i O2.
Uzyskane rezultaty nale1y poda+ w formie widm ró1nicowych (po odj.ciu widma podstawowego). Na podstawie danych zawartych w poni1szej tabeli zostanie przeprowadzona analiza uzyskanych widm i zaproponowany zostanie przebieg reakcji NO+C3H6+O2.
Zestawienie pasm wraz z odpowiadaj cymi im zaadsorbowanymi grupami:
Cz-stotliwo</ po adsorpcji NO Grupa
~1905cm-1 NO zaadsorbowany na centrach Cu2+ ~1874cm-1 gazowy NO 1860cm-1 Formy Cu+-(NO)2. 1630-1660cm-1 azotany(V), gazowy NO 2 1400-1500cm-1 zaadsorbowane cz steczki NO2 pasma poni1ej 1350cm-1 NO3-i NO2
-Cz-stotliwo</ po adsorpcji HC Grupa
1530, 1461 i 1346cm-1 –COOH i –COH
dublet przy 1464 i 1442cm-1, pasmo przy
1365cm-1 ugrupowania -CH3
1340cm-1 –CH
1422cm-1 =CH2
~1670cm-1 C=C
4
Temperaturowo programowana redukcja
wodorem (TPR-H
2) w badaniach
oddzia ywa faza aktywna-no<nik
Literatura uzupe niaj ca:Barbara Grzybowska-Owierkosz, „Elementy Katalizy Heterogenicznej”, PWN, Warszawa 1993, str. 143-144
1. Wst-p
Metoda TPR zosta a po raz pierwszy opisana w 1975 roku przez Robertsona i wspó pracowników w Journal of Catalysis – rys. 1.
Metoda ta polega na redukcji próbki w mieszaninie wodoru rozcie,czonego gazem oboj.tnym (najcz.%ciej argonem) podczas liniowego narostu temperatury (najcz.%ciej 10K/min). W chwili rozpocz.cia redukcji zu1ywany jest wodór, a przewodnictwo cieplne mieszaniny ulega zmianie, co rejestrowane jest przez katarometr.
Rysunek 1. Tytu owa strona historycznej publikacji Robertsona.
Koncepcja metody TPR przedstawiona zosta a na rys. 2. W procesie redukcji ilo%+ redukowalnej substancji (n) b.dzie si. zmniejsza a do pewnego momentu spadaj c do zera ze wzrostem temperatury. Jednocze%nie, stopie, redukcji b.dzie wzrasta od zera do finalnej, najwy1szej warto%ci =1. Pierwsza pochodna tej krzywej , dn/dt, daje krzyw redukcji przechodz c przez maksimum.
Szybko%+ redukcji jest proporcjonalna do szybko%ci konsumpcji wodoru, która mo1e by+ monitorowana w funkcji czasu.
Najcz.%ciej stosowane mieszanki redukuj ce to: a). 5-10% H2/Ar
b). 5-10% H2/N2
c). 5% CO/Ar
Na rysunku 3 przedstawiono typow aparatur. do bada, TPR. Sk ada si. ona z nast.puj cych elementów:
- reaktor - piec - programator temperatury - detektor cieplno-przewodno%ciowy (TCD) - wymra1alnik gdzie: - stopie, redukcji dn/td – szybko%+ redukcji T – temperatura t – czas n – ilo%+ redukowalnej substancji
Gaz przep ywa przez reaktor a nast.pnie przez wymra1alnik, w którym kondensuje si. woda b.d ca produktem redukcji. Obj.to%+ uk adu pomi.dzy reaktorem a detektorem powinna by+ mo1liwie jak najmniejsza, aby unikn + rozmywania pików. Wymagana jest te1 dodatkowa linia gazowa s u1 ca wst.pnej aktywacji próbki przed w a%ciwym pomiarem TPR (najcz.%ciej stosuje si.: tlen, powietrze, gaz oboj.tny). Sygna detektora jest rejestrowany wraz z temperatur mierzon w z o1u katalizatora.
Uk ad powy1szy, o ile wykonany jest z odpowiednich materia ów i zaopatrzony w kilka dodatkowych linii gazowych, s u1y+ mo1e tak1e jako aparat do Temperaturowo Programowanego Utleniania (TPO), Temperaturowo Programowanej Desorpcji (TPD) czy Temperaturowo Programowanego Siarczkowania (TPS).
Gazy stosowane w pomiarach TPR musz charakteryzowa+ si. wysok czysto%ci szczególnie, je%li chodzi o zawarto%+ tlenu i wody. W przeciwnym przypadku konieczne jest stosowanie odtleniaczy i odwadniaczy. Niektórzy autorzy zalecaj stosowanie odtleniaczy i odwadniaczy nawet dla gazów o wysokiej czysto%ci.
2. Warunki prowadzenia procesu TPR
W celu uzyskania maksymalnej czu o%ci metody, ró1nica st.1e, wodoru przed i za reaktorem musi by+ maksymalnie du1a.
O czu o%ci metody decyduje wiele czynników: - szybko%+ grzania (K/s)
- pocz tkowa ilo%+ redukuj cej si. substancji n0(µmol)
- szybko%+ przep ywu gazu F (cm3/s)
- st.1enie H2w gazie redukuj cym c0(µmol/cm3)
Zdefiniowano parametr K, który wi 1e te parametry i pozwala na wybranie optymalnych
warunków pomiaru. Dla szybko%ci narostu temperatury pomi.dzy 0,1 a 0,3 K/s (6-18 K/min) parametr ten okre%lony jest wzorem:
0 0
c
F
n
K
×
=
Aby uzyska+ optymalny profil TPR warto%+ parametru K musi zawiera+ si. w przedziale pomi.dzy 55s a 140s. Dla K mniejszego od 55s, czu o%+ metody staje si. zbyt niska; dla K powy1ej 140s konsumpcja wodoru jest zbyt du1a.
Uwzgl.dniaj c szybko%+ narostu temperatury mo1emy zaproponowa+ wzór w postaci:
0 0
c
F
n
P
×
×
=
Dla uzyskania optymalnego profilu TPR warto%+ tego parametru powinna by+ mniejsza lub równa 20K.
Prze%ledVmy zatem, w jaki sposób warunki eksperymentu wp ywaj na obraz profili TPR. Na rys.4.A mo1emy obserwowa+ wp yw szybko%ci narostu temperatury na profile TPR – na po o1enie maksimum piku oraz na szeroko%+ piku. Jak widzimy, wzrost szybko%ci
narostu temperatury powoduje, 1e maksimum piku przesuwa si. wyraVnie w kierunku wy1szych temperatur. Przesuni.cie to wynosi 75oC w przypadku szybko%ci grzania 1K/min a
30K/min. Bior c pod uwag. najcz.%ciej stosowane szybko%ci narostu temperatury ró1nice te wynosz oko o 20K dla szybko%ci 10 i 20K/min.
Na rys.4.B. przedstawiono wp yw st.1enia H2 w mieszaninie redukuj cej na po o1enie profili
TPR. Mniejsze st.1enie H2 powoduje przesuni.cie maksimum piku w kierunku wy1szych
temperatur. Dla najcz.%ciej stosowanego zakresu st.1e, wodoru (5-10% obj.) ró1nica w po o1eniu maksimów pików wynosi oko o 15K.
Najmniejszy wp yw na po o1enie maksimum piku i kszta t profilu TPR ma szybko%+ przep ywu gazu redukuj cego rysunek 4C.
Rysunek 4D obrazuje wp yw ilo%ci substancji redukowalnej na profil TPR i po o1enie maksimum piku. Oczywistym jest, 1e im wi.cej substancji redukujemy – tym otrzymany sygna ma wi.ksz intensywno%+. Obserwuje si. tak1e ze wzrostem nawa1ki stopniowe przesuwanie temperatury maksimum piku w kierunku wy1szych warto%ci.
Wp yw wyszczególnionych czynników decyduj cych o kszta cie i po o1eniu profili TPR zosta przeanalizowany na podstawie modelu matematycznego. Nale1y zaznaczy+, 1e model ten doskonale sprawdza si. dla uk adów rzeczywistych.
3. Zastosowanie techniki TPR
Metod. Temperaturowo Programowanej Redukcji mo1emy wykorzystywa+ w badaniach ka1dej substancji, która ulega redukcji. Spectrum mo1liwo%ci jest tutaj ogromne – jedynym ograniczeniem jest wyobraVnia eksperymentatora.
Technika TPR najcz.%ciej wykorzystywana jest w nast.puj cych badaniach:
- redukowalno%ci czystych tlenków metali
- redukowalno%ci uk adów zawieraj cych dwa lub wi.cej tlenków - tlenków metali osadzonych na no%nikach
- oddzia ywa, fazy aktywnej z no%nikiem
- oddzia ywa, prekursorów fazy aktywnej z no%nikiem
- identyfikacji kompleksów powierzchniowych na katalizatorach sta ych - depozytów w.glowych
Prze led4my zatem rzeczywiste przyk ady profili TPR ró$nych substancji. a) Czyste tlenki metali
Jako przyk ad pos u1 nam tlenki miedzi i uk ad bitlenkowy CuO-ZnO. Tlenki miedzi (Cu2O i CuO) daj (przy zachowaniu w a%ciwych warunków pomiaru) jeden ostry pik na
profilu TPR – rysunek 5A i B. Wyniki te sugeruj , 1e CuO redukuje si. do metalu w jednym etapie zgodnie z równaniem reakcji:
CuO + H2 Cu0 + H2O
W podobny sposób redukuje si. CuO w uk adzie z ZnO (stosunek molowy CuO:ZnO = 67:33). Ilo%+ zu1ytego wodoru wskazuje na ca kowit redukcj. CuO do Cu0. Maksimum piku
redukcyjnego przypada na temperatur. 480K, czyli ok. 24K ni1sz ni1 dla czystego CuO. Obni1enie temperatury redukcji CuO dla uk adu CuO-ZnO %wiadczy o wi.kszej redukowalno%ci CuO w tym uk adzie w porównaniu z czystym CuO. Spowodowane mo1e to by+ np. wi.kszym rozdrobnieniem tlenku miedzi w uk adzie CuO-ZnO.
W pewnych sytuacjach, gdy warunki eksperymentu nie s w a%ciwie dobrane (niew a%ciwa warto%+ parametru K), na krzywej TPR pojawiaj si. artefakty w postaci drugiego piku, co niekiedy jest b .dnie przypisywane etapowej redukcji CuO do Cu2O a
nast.pnie do Cu0. Widzimy to na rysunku 6.
Rysunek 5. Profile TPR dla:
A – Cu2O,
B – CuO, C – CuO-ZnO.
Rysunek 6. Artefakt na krzywej TPR tlenku