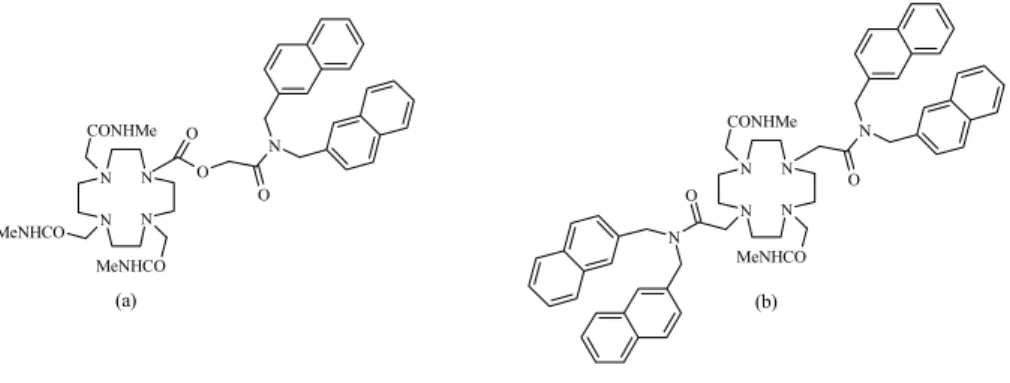
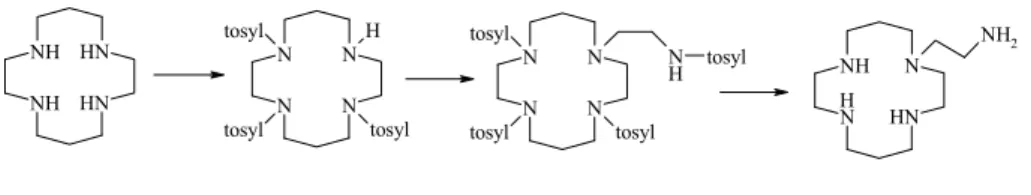
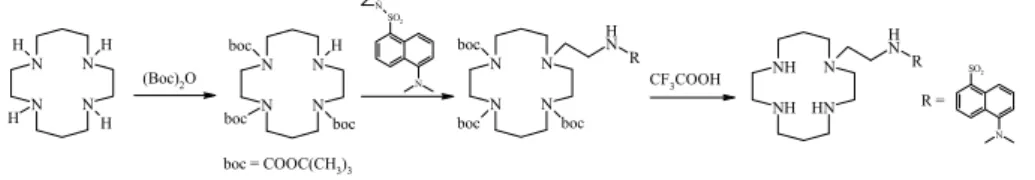
Syntetyczne receptory jonowe
Pod
redakcją Grzegorza Schroedera
SERIA: Chemia Supramolekularna
Recenzent : Dr hab. Jan Milecki
Prof. dr hab. Grzegorz Schroeder Uniwersytet im. A. Mickiewicza Wydział Chemii Grunwaldzka 6 60-780 Poznań tel.: 0-61-8291-486 E-mail: schroede@amu.edu.pl ISBN 83-89936-05-4 „BETAGRAF”P.U.H. Poznań
SPIS TREŚCI
1. Syntetyczne receptory jonowe - jonofory
Grzegorz Schroeder, Błażej Gierczyk... 4
2. Spektrometrii mas - technika elektrorozpylania (ESIMS)
Nataliya Lyapchenko, Grzegorz Schroeder ...34
2. Wybrane zagadnienia katalizy międzyfazowej
Bogusława Łęska ...67
4. Synteza i właściwości pochodnych makrocyklicznych
poliamin
1. Syntetyczne receptory jonowe - jonofory
Grzegorz Schroeder, Błażej Gierczyk
Receptory molekularne to układy zdolne do wiązania innych cząsteczek lub jonów w aktywnych fragmentach swojej struktury na zasadzie kompleksów gość-gospodarz. Zdolność związków do wiązania z receptorem jest uzależniona od powinowactwa i aktywności wewnętrznej. Powinowactwo zależy od stopnia dopasowania struktury cząsteczki związku (gościa) do miejsc wiążących receptora (gospodarza). Im jest ono większe, tym bardziej swoiste i trwałe jest związanie gościa z receptorem i ukierunkowane działania chemiczne kompleksu (metoda klucza i zamka). Niewielka różnica w konfiguracji atomów w cząsteczce lub jej wielkości mogą osłabiać, zmniejszyć wybiórczość i siłę działania. Aktywność wewnętrzna określa zdolność do pobudzenia receptora i wyzwalania reakcji biologicznej. Związki chemiczne o dużym powinowactwie do receptora i dużej aktywności wewnętrznej, które łatwo wiążą się z receptorem i wywołują zaprogramowaną dla danego receptora odpowiedź biologiczną, nazywane są agonistami. Związki, które wiążą się z receptorem nie wywołując jego reakcji a zatem odpowiedzi biologicznej nazywamy antagonistami. Utrudniają one dostęp do receptora przez substancje agonistyczne, konkurując z nimi o miejsca receptorowe. Trwałość połączeń związek-receptor, zależy od rodzaju oddziaływań pomiędzy związkiem chemicznym i receptorem.
W grupie receptorów molekularnych szczególne miejsce zajmują jonofory. Jonofory to hydrofobowe cząsteczki zdolne do transportu jonów z fazy wodnej do hydrofobowej warstwy, lub zdolne do transportu jonów poprzez warstwę lipidową do komórki. Jonofory definiuje się często jako nośniki, akceptory lub jako cząsteczki gospodarza dla jonów Na+, K+, czy Ca2+, czyli
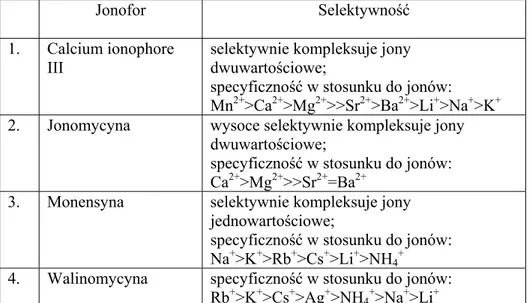
takich, które są transportowane poprzez membrany biologiczne. Selektywność jonoforu w stosunku do kationów litowców i berylowców wynika zarówno z jego trójwymiarowej struktury, jak i różnicy energii desolwatacji i kompleksowania. Czynnikami decydującymi o selektywności jonoforu są przede wszystkim: polarność wnęki, lipofilowość powierzchni, wiązania wodorowe utrzymujące strukturę oraz oddziaływania jon-dipol. Znaczącą rolę w procesie selektywnego rozpoznawania molekularnego odgrywa także pH środowiska, siła jonowa oraz typ rozpuszczalnika. Tabela 1 przedstawia kilka przykładów selektywności jonoforów.
Tabela 1. Selektywność wybranych jonoforów
Jonofor Selektywność
1. Calcium ionophore III
selektywnie kompleksuje jony dwuwartościowe;
specyficzność w stosunku do jonów: Mn2+>Ca2+>Mg2+>>Sr2+>Ba2+>Li+>Na+>K+ 2. Jonomycyna wysoce selektywnie kompleksuje jony
dwuwartościowe;
specyficzność w stosunku do jonów: Ca2+>Mg2+>>Sr2+=Ba2+
3. Monensyna selektywnie kompleksuje jony jednowartościowe;
specyficzność w stosunku do jonów: Na+>K+>Rb+>Cs+>Li+>NH
4+
4. Walinomycyna specyficzność w stosunku do jonów: Rb+>K+>Cs+>Ag+>NH4+>Na+>Li+
Walinomycyna charakteryzuje się 10 000 razy większym powinowactwem do jonów potasu niż jonów sodu.
Istnieje kilka klas naturalnych acyklicznych i cyklicznych jonoforów1,2. Chemicy opanowali metody syntezy związków wykazujących
właściwości kompleksujące w stosunku do jonów metali 1 i 2 grupy układu okresowego. Syntetyczne jonofory na trwałe wpisały się w współczesną
chemię, biologię czy ochronę środowiska. Stanowią bazę do budowy wielu biomolekularnych urządzeń, których zastosowanie rośnie lawinowo z roku na rok. 3,4,5
W niniejszym opracowaniu przedstawiono zestawienie syntetycznych jonoforów dostępnych w handlu z zachowaniem powszechnie stosowanych nazw angielskich lub skrótów.
I. Jonofory dla jonów H+6
1. Hydrogen ionophore I
N
2. Hydrogen ionophore II, ETH 1907
N
3. Hydrogen ionophore III7
N
4. Hydrogen ionophore IV, ETH 1778 8
N
O O
II. Jonofory dla jonów amonu (NH4+)
1. Ammonium ionophore I; Nonactin9
O O O O O O O O O O O O
III. Jonofory dla jonów baru (Ba2+)
1. Barium ionophore I10 O O O O O N N
IV. Jonofory dla jonów cezu (Cs+)
1. Cesium ionophore I11
O
O O
2. Cesium ionophore II
O O O
6
V. Jonofory dla jonów kadmu (Cd2+)
1. Cadmium ionophore I12 O S O S N N
VI. Jonofory dla jonów litu (Li+)
1. Lithium ionophore I, ETH 14913
O O
O O
N
2. Lithium ionophore II, ETH 164414
O
O
N
N
3. Lithium ionophore III, ETH 181015
O
O
N
N
4. Lithium ionophore IV, ETH 213716
N N
O O
O O
5. Lithium ionophore V, 12-Crown-417
O O
O O
6. Lithium ionophore VI, 6,6-Dibenzyl-14-crown-418
O O O O
7. Lithium ionophore VII19 O O O O O P O O O
8. Lithium ionophore VIII20
N N N O O O O O O
VII. Jonofory dla jonów magnezu (Mg2+)
1. Magnesium ionophore I, ETH 111721
O O N N
2. Magnesium ionophore II22 N H N H O O O O N N
3. Magnesium ionophore III23
N H N H O O O O N N
4. Magnesium ionophore IV, ETH 702524
N NH N N H O O O O N O O N 5. Magnesium ionophore V25 O N O O N O N H O O N H O O
6. Magnesium ionophore VI, ETH 550626
N H N H NH O O N O N O O O N
VIII. Jonofory dla jonów miedzi (Cu2+)
1. Cooper ionophore I, o-XBDiBBDTC27
S N
S N
S S
IX. Jonofory dla jonów ołowiu (Pb2+)
1. Lead ionophore I, ETH 32228
O O O O N N
2. Lead ionophore II, MBDiBDTC29
S S
S N
S
3. Lead ionophore III, ETH 543530 N O S O S N 4. Lead ionophore IV31 O S N 4
5. Lead ionophore V, 15-Crown-532
O O O
O O
X. Jonofory dla jonów potasu (K+)
1. Potassium ionophore I, Walinomycin33
O N H O N H O O O O 3 2. Potassium ionophore II34 O O O O O O O O O O O O O O O O
3. Potassium ionophore III, BME 4435
O O O O O O N H O O O2N O O O O O O NH O O NO2
4. Potassium ionophore IV36 O O O O O O O 2 5. PBFI(AM) O O O O O O O O O O O O O O O O O O O O O N O O N O
XI. Jonofory dla jonów srebra (Ag+)
1. Silver ionophore II, MAO37
O S S
2. Silver ionophore III38 S S S N S N 3. Silver ionophore IV39 O S OH 2
XII. Jonofory dla jonów sodu (Na+)
1. Sodium ionophore I, ETH 22740
O O O O N N O N O
2. Sodium ionophore II, ETH 15741 O O O O N N
3. Sodium ionophore III, ETH 212042
O O O O N N 4. Sodium ionophore IV43 O O O O O
5. Sodium ionophore V, ETH 412044 O O O O N N O O 6. Sodium ionophore VI45 O O O O O O O O O O O O
7. Sodium ionophore VII O O O O O O O O O O O O 8. Sodium ionophore X46 O O O 4 9. SBFI (AM)47 O O O O O O O O O O O N N O O O O O O O O O O O O
10. SBFI48 O O O N N O O O O COOH COOH HOOC COOH 11. SQI-Pr O O O O N N O N N O O 12. SQI-Et O O O O N N O N N O O
XIII. Jonofory dla jonów UO22+ 1. Uranyl ionophore I48 O O O O N N
XIV. Jonofory dla jonów wapnia (Ca2+)
1. Calcium ionophore I, ETH 100149
O O O O N N O O O O
2. Calcium ionophore II, ETH 12950
N O N
O O
3. Calcium ionophore III, Calcium ionophore A23187, Calcimycin A2318751 N O NH O O O N H O OH
4. Calcium ionophore IV, ETH 523452 N O N O O
5. Calcium ionophore V, K23E153
O O N H O O N O O O N O O O N H O 6. 4-Br A23187 N O NH O O O N H O OH Br 6. FURA-PE3 (AM) N O O O N O O O O O O O O N N N O O O O O O O O O O O O O O O O O O
7. FURA-PE3 N O O N N N N O O O HOOC HOOC COOH COOH COOH HOOC 8. INDO-PE3 (AM) N O N O O O O O O O O N N N O O O O O O O O O O O O O O O O O O 9. INDO-PE3 N N N N N O O O HOOC HOOC COOH COOH COOH COOH
10. FFP18 N O O N N N N O O O HOOC HOOC COOH COOH HOOC 11. FIP18 N N N N N O O O HOOC HOOC COOH COOH COOH 12. FURA-2FF N O O N N O O HOOC HOOC COOH COOH HOOC F F
XV. Jonofory dla anionów CO32-, RCOO- i OH
-1. Carbonate ionophore I, ETH 601054
CF3 O
O O
2. Carbonate ionophore II, ETH 601955
CF3 O
S O O
3. Carbonate ionophore III, ETH 602256
CF3 O
N O
4. Carbonate ionophore IV57
F3C O
XVI. Jonofory dla jonów Cl
-1. Chloride ionophore I58 N N N N Mn Cl 2. Chloride ionophore II59 Hg Hg O O O CF3 CF3 O O O
3. Chloride ionophore III60 Hg Hg O O Cl Cl
XVII. Jonofory dla jonów HSO3
-1. Hydrogen sulfite ionophore I, ETH 544461
O O
OHC
XVIII. Jonofory dla jonów NO2
-1. Nitrite ionophore I62 N N N N Co+ O O O O O O O O O O O O O O CN OH2 ClO4
-1. Nitrite ionophore II63 N N N N Co O O O O O O O O O O O O O O CN CN
2. Nitrite ionophore III64
N N N N Co O NH2 NH2 O O N H2 N H2 O O N H2 N H2 O O N H2 CN OH2
XIX. Jonofory dla jonów SO4 2-1. Sulfate ionophore65 S N H N H N H N H S
XX. Jonofory dla amin I-rzędowych
1. Amine ionophore I66
O O O
6
Literatura
1. W. Burgermeister, R. Winkler-Oswatitsch, Topics in Current Chem., 69, 91, 1977
2. R. Hilgenfeld, W. Saenger, Top. Curr. Chem.,101, 1, 1982
3. T.Schrader, A.D.Hamilton, Functional Synthetic Receptors, Wiley-VCH, Weinheim, 2005
4. I.Willner, E.Katz, Bioelectronics, From Theory to Applicacations, Wiley-VCH, Weinheim, 2005;
5. J. Aidley, P.R.Stanfield, Ion Channels Molecules in Action, University Press, Cambridge, 2003).
6. P. Chao et al., Europ. J. Physiol., 411, 216, 1988 7. L. Wu et al., Talanta, 34, 577, 1987
8. U. Oesch et al., Med. Biol. Eng. Comput., 25, 414, 1987; U. Oesch et al., Anal. Chem., 58, 2285, 1986
9. Y. M. Fraticell et al., Anal. Chem., 53, 992, 1981; B. C. Pressman,
Ann. Rev. Biochem., 45, 501, 1976; . F. Morf et al., Helv. Chim. Acta, 54, 2863, 1971; W. Simon, Pure Appl. Chem., 25, 811, 1971;
F. Eisenman et al., Ann. N.Y. Acad. Sci., 264, 34, 1975; D. Ammann,
Ion-Selective Rev., 5, 3, 1983; W. Keller-Schierlen et al., Fortsch. Chem. Org. Naturstoffe, 26, 161, 1968
10. M. W. Laubli, et al., Anal. Chem., 57, 2756, 1985 11. A.Cadogan et al., Analyst, 115, 1207, 1990
12. K. Schneider et al., Helv. Chim. Acta, 63, 217, 1980
13. D. Ammand et al., Ion and Enzyme Electrodes in Biology and
Medicine, 1976; W. Simon et al., Methods Enzymol., 56, 439, 1979
14. D. Erne et al., Helv. Chim. Acta, 65, 538, 1982; A. F. Zhukow et al.,
Anal. Chem. Acta, 131, 177, 1981
15. E. Metzger et al., Anal. Chem., 59, 1600, 1987 16. E. Metzger et al., Anal. Chem., 59, 1600, 1987 17. V. P. Y. Gadzepko et al., Anal. Lett., 16, 1371, 1983 18. K. Kimura et al., Anal. Chem., 59, 2331, 1987 19. K. Kimura et al., J. Chem. Soc. Perkin II, 1945, 1986
20. M. Bocheńska et al., J. Inclusion Phenom. Recogn. Chem., 10, 19, 1991
21. F. Lanter et al., Anal. Chem., 52, 2400, 1980; D. Erne et al., Helv.
Chim. Acta, 63, 2271, 1980
22. Z. Hu et al., Anal. Chem., 61, 574, 1989
23. A.Muller et al., Mikrochim. Acta, III, 283, 1988; M. Mai-Żurawska et al., Anal. Chim. Acta., 218, 47, 1989
24. E. Spichiger et al., Frasenius J. Anal. Chem., 341, 727, 1991 25. K. Suzuki et al., Anal. Chem., 67, 324, 1995
26. J. O’Donnell et al., Anal. Chim. Acta, 281, 129, 1993; U. E. Spichiger et al., Electrichim. Acta, 42, 3137, 1997
27. S. Kamata et al., Analyst, 114, 1029, 1989 28. E. Linder et al., Anal. Chem., 56, 1127, 1984 29. S. Kamata et al., Anal. Chem., 63, 1995, 1991 30. M. Lerchi et al., Anal. Chem., 64, 1534, 1992
31. E. Malinowski et al., Anal. Chim. Acta, 298, 253, 1994 32. S. K. Srivastava et al., Analyst, 120, 495, 1995
33. A. D. Bangham et al., Chem. Phys. Lip., 8, 386, 1972; L. A. R. Pioda et al., Clin. Chim. Acta, 29, 289, 1970; H. F. Osswald et al., Clin.
Chem., 25, 39, 1979
34. H. Tamura et al., Microchim. Acta II, 287, 1983; K. Kimura et al., J.
Electroanal. Chem., 95, 91, 1979
36. A. Cadogan et al., Anal. Proc., 28, 13, 1991
37. J. Casabo et al., J. Chem. Soc. Dalton Trans., 1969, 1991 38. M. Lerchi et al., Anal. Chem., 66, 1713, 1994
39. D. N. Reinhoudt et al., Anal. Chim. Acta, 298, 245, 1994
40. R. A. Steiner et al., Anal. Chem., 51, 351, 1979; W. Simon et al.,
Methods Enzymol., 56, 439, 1979
41. W. Simon et al., Pure Appl. Chem., 44, 613, 1975; D. Ammann et al., Ion and Enzyme Electrodes in Biology and Medicine, 1976; W. Simon et al., Methods Enzymol., 56, 439, 1979
42. T. Maruizumi et al., Microchim. Acta, I, 331,1986 43. K. Suzuki et al., Anal. Chem., 68, 208, 1996 44. T. Gehrig et al., Anal. Chim. Acta, 233, 295, 1990
45. H. Tamura et al., Anal. Chem., 54, 1224, 1982; D. J. Cram et al.,
Science, 183, 803, 1974; T. Shono, J. Electroanal. Chem., 132, 99,
1982
46. D. Diamond et al., Analyst, 114, 1551, 1989; D. Diamond et al.,
Anal. Chem., 64, 2496, 1992
47. A. Minta et al., J. Biol. Chem., 264, 19449, 1989
48. J. Senkyr et al., Anal. Chem., 51, 786, 1979; P. A. Bertrand et al,
Anal. Chem., 55, 364, 1983; E. Malinowska, Analyst, 115, 1085,
1990
49. W. Simon et al., Ann. N. Y. Acad. Sci., 307, 52, 1978; P. Caroni et al., Biochem. Biophys. Acta, 470, 437, 1977; W. Simon et al.,
Methods in Enzymology, 56, 439, 1979
50. E. Pretsch et al., Helv. Chim. Acta, 63, 191, 1980; U. Schefer et al.,
Anal. Chem., 58, 2282, 1986
51. R. F. Kauffman et al., J.Biol. Chem., 255, 2735, 1980; H. Markram et al., Brain Res., 540, 322, 1991; B. C. Pressman et al., Ann. Rev.
Biochem., 45, 50, 1976; R. G. Knowles et al., Trends Biochem. Sci.,
17, 399, 1992
52. P. Gehrig et al., Chimia, 43, 377, 1989
53. H. Hisamoto et al., Anal. Chim. Acta, 299, 179, 1994; K. Suzuki et al., Anal. Chem., 67, 324, 1995
54. K. Wang et al., Anal. Chem., 63, 970, 1991; C. Behringer et al.,
Anal. Chim. Acta, 233, 41, 1990
55. C.Behringer et al., Anal. Chim. Acta, 233, 41, 1990; K. Wang et al.,
Anal. Chem., 63, 970, 1991
56. C.Behringer et al., Anal. Chim. Acta, 233, 41, 1990; K. Seiler et al.,
Anal. Chim. Acta, 244, 151, 1991; U. E. Springer et al., Biosensors, Bioelec., 7, 715, 1992
57. M. E. Meyerhoff et al., Anal. Chem., 59, 144, 1987; O. Dinten et al.,
Anal. Chem., 63, 596, 1991
59. M. Rothmaier et al., Anal. Chim. Acta, 271, 135, 1993 60. M. Rothmaier et al., Anal. Chim. Acta, 327, 17, 1996 61. M. Kuratli et al., Anal. Chem., 66, 85, 1994
62. R. Stepanek et al., Anal. Chim. Acta, 182, 83, 1986
63. S. Daunert et al., Prog. Clin. Biol. Res., 292, 215, 1989; S. A. O’Reilly et al., Anal. Chem., 63, 1278, 1991
64. L. G. Bachas et al., Anal. Chim. Acta, 256, 269, 1992 65. S. Nishizama et al., Anal. Chim. Acta, 358, 35, 1998 66. X. H. Gu et al., Analyst, 118, 863, 1993
2. Spektrometrii mas - technika elektrorozpylania
(ESIMS)
Nataliya Lyapchenko, Grzegorz Schroeder
Elektrorozpylanie jest złożonym procesem, w trakcie którego silne pole elektryczne rozprasza ciekłą próbkę do fazy gazowej, z utworzeniem aerozolu składającego się z bardzo drobnych, wysoce naładowanych kropli, z których po odparowaniu rozpuszczalnika powstają jony w fazie gazowej[1].
Elektrorozpylanie (ang. electrospray, ES) jest zatem techniką pozwalającą na przeniesienie jonów z roztworu do fazy gazowej.
Obecność naładowanych kropli odgrywa znaczącą rolę w zjawiskach atmosferycznych: naładowane krople tworzące chmury uczestniczą w tworzeniu błyskawic[2-4], wysoce naładowane nanokrople powodują nadmierną absorpcję promieniowania słonecznego, a z kolei promieniowanie radioaktywne wzmacnia naładowanie aerozoli w środowisku[2].
Już w osiemnastym wieku znane było zjawisko elektrostatycznego rozpylania cieczy, czyli wytworzenia aerozolu poprzez przyłożenie wysokiego napięcia do cieczy przepuszczanej pod wysokim ciśnieniem przez cienką kapilarę[5]. Na początku XX wieku próbę badania zjawiska
rozpylania cieczy pod wpływem pola elektrycznego podjął Zeleny[6-8].
Zbadał on:
- warunki tworzenia i rozpraszania naładowanych kropli z obojętnego roztworu, przepuszczanego przez naelektryzowaną kapilarę;
- wpływ na ten proces: napięcia przykładanego do kapilary, średnicy kapilary, promieniowania β i γ, rodzaju rozpuszczalnika, rodzaju materiału, z którego wykonana jest końcówka kapilary oraz rodzaju gazu otaczającego kapilarę;
- charakterystyki rozładowywania elektrycznego kropli oraz warunki powstania areozolu i różnice między rozpylaniem jonów dodatnich a ujemnych.
Badania te miały charakter obserwacyjno-jakościowy z wykorzystaniem niskorozdzielczych zdjęć fotograficznych, bez pomiaru średnicy i ładunku kroplel[9].
Obecnie zjawisko elektrorozpylania znalazło zastosowanie w: drukowaniu i malowaniu, wytwarzaniu pyłu metalicznego i koloidów, opryskiwaniu plonów oraz do wytwarzania gazu pędnego w tak zwanych rakietach jonowych[3,5,10].
Do zastosowania elektrorozpylania w spektrometrii mas doszło na skutek prac Dole’a i wsp., którzy pod koniec lat 60-tych XX wieku podjeli próbę uzyskania w próżni strumienia makrojonowego[5,10].
Zasada zastosowania elektrorozpylania w spektrometrii mas polega na tym, że rozcieńczony roztwór analitu przechodzi przez cienka, ogrzewaną kapilarę, do której przykłada się potencjał rzędu 2 – 10 kV [10]. Roztwór ten
jest następnie elektrostatycznie rozpylany do komory jonizacyjnej, wypełnionej gazem, w wyniku czego tworzy się aerozol naładowanych kropli. System kolejnych pomp próżniowych wytwarza warunki niezbędne do detekcji cząsteczek metodą spektrometrii mas, a elektrostatyczny układ ogniskujący kieruje jony do analizatora mas. Gdy kapilara względem ścianek komory jonizacyjnej jest naładowana dodatnio, mówi się o przeprowadzaniu elektrorozpylania w trybie jonów dodatnich; w przeciwnym przypadku mówi się o trybie jonów ujemnych.
O wiodącym miejscu metody elektrorozpylania spośród innych technik spektrometrii mas decydują trzy cechy[5]:
1. wyjątkowa zdolność tworzenia jonów wielokrotnie naładowanych, pozwalająca na obserwację makrocząsteczek za pomocą dostępnych analizatorów o średnich zakresach m/z;
2. możliwość wprowadzenia próbki w postaci roztworu, co jest niezmiernie ważne, przy badaniu próbek pochodzenia biologicznego i środowiskowego, w badaniach kompleksów, asocjatów oraz procesów agregacji;
3. nadzwyczajna „miękkość” metody, pozwalająca na zachowanie w fazie gazowej słabych oddziaływań międzycząsteczkowych istniejących w roztworze.
Rodzaj cząsteczek, które mogą być analizowane z użyciem metody ESIMS, waha się od: soli organicznych i nieorganicznych, istniejących w roztworze w postaci zjonizowanej, przez polarne substancje obojętne, które asocjacjują, substancje niepolarne, które w kapilarze mogą ulegać elektrochemicznemu utlenieniu/redukcji aż po małe jony, takie jak proton, jony litu czy sódu[11].
1. Tworzenie jonów w fazie gazowej
Proces utworzenia jonów w fazie gazowej podczas procesu elektrorozpylania schematycznie został przedstawiony na rysunku 1.
Proces tworzenie jonów w fazie gazowej z roztworu zawiera trzy zasadnicze etapy[5,12]:
(a) utworzenie naładowanych kropli przy końcówce kapilary (zaznaczone jako (a) na rys. 1);
(b) kurczenie się naładowanych kropli przy odparowywaniu rozpuszczalnika oraz utworzenie kropli wtórnych (następczych), będących bezpośrednim źródłem jonów w fazie gazowej (zaznaczone jako (b) na rys. 1);
(c) utworzenie jonów w fazie gazowej (zaznaczone jako (c) na rys. 1).
Tworzenie naładowanych kropli przy końcówce kapilary ES
Na skutek oddziaływania pola elektrycznego na roztwór analitu dochodzi do częściowego rozdzielenia jonów: ulegają one ruchowi
elektroforetycznemu odpowiednio do przyłożonego pola elektrycznego [9,11,13-15]. W trybie jonów dodatnich aniony migrują w kierunku metalowych
ścianek kapilary ES, tymczasem kationy migrują w kierunku od kapilary do elektrody przeciwnie naładowanej. W trybie jonów ujemnych, kierunek elektroforetycznego ruchu kationów i anionów jest przeciwny, niż w przypadku trybu jonów dodatnich.
utlenianie redukcja + _ elektrony elektrony źródło napięcia kapilara
elektroda przeciwnie naładowana
+ + + + + + - -- -+ + + -+ + ++ -+ ++ + + + + + + + + -+ + + + + + + + + + + (a) (b) (c) +
Rys. 1. Schemat procesu elektrorozpylania (tryb jonów dodatnich): (a) – utworzenie naładowanych kropli, (b) – kurczenie się naładowanych kropli z utworzeniem kropli wtórnych, (c) – utworzenie jonów w fazie gazowej. W wyniku takiego rozdzielenia jonów, blisko końcówki kapilary kumuluje się nadmiar ładunku, a przyciąganie jego do elektrody przeciwnej powoduje wydłużenie wychodzącej kropli w kierunku tej elektrody. Siła pola elektrycznego, wypychająca roztwór jonów równoimiennych, jest równoważona siłą napięcia powierzchniowego cieczy[1,11,13]. Skutkiem tej
konkurencji pomiędzy napięciem powierzchniowym cieczy, utrzymującym ją w całości, a oddziaływaniem jonów z polem elektrycznym, powodującym destabilizację roztworu, jest nadanie cieczy formy dynamicznego stożka (stożek Taylora1, ang. Taylor cone)[1,11,13], którego końcówka jest jego
najmniej stabilnym punktem ze względu na najwyższą w tym miejscu gęstość ładunku[1]. W przypadku przyłożenia do kapilary odpowiednio
wysokiego napięcia, gęstość ładunku przybiera na końcówce stożka wartość na tyle wysoką, że napięcie powierzchniowe nie może utrzymywać stożka cieczy w całości, skutkiem tego jest wydłużenie stożka do postaci nitki, która rozpada się na strumień drobnych równoimiennie naładowanych kropli. Elektrostatyczne odpychanie takich kropli rozprasza strumień do postaci spreju, w którym wyodrębnia się rdzeń, składający się z kropli o równej wielkości, i otaczającą mgłę, składającą się z mniejszych kropli towarzyszących[9]. Utworzona w ten sposób chmura drobnych,
naładowanych kropli migruje w kierunku elektrody przeciwnie naładowanej, głównie pod wpływem potencjału pola elektrycznego oraz w mniejszym stopniu pod wpływem gradientu ciśnienia[11,13].
Kurczenie się naładowanych kropel przy odparowywaniu rozpuszczalnika
Na kurczenie się naładowanych kropel wpływają różne procesy fizykochemiczne[2,11,16]. Podczas rozprzestrzeniania się aerozolu naładowanych kropel w komorze jonizacyjnej dochodzi do kolizji kropel z
1 Określenie „stożek Taylora” oraz teoretyczna analiza zachowania tego stożka w polu elektrycznym przeprowadzona przez Taylora dotyczą cieczy statycznej. Dla takiej cieczy stożkowa forma menisku oraz jej odnawianie poprzez cykl procesów odrywania kropli i uwypuklenia następnej porcji cieczy wynika z wahań pomiędzy siłami napięcia powierzchniowego a siłami oddziaływania pola elektrycznego z dipolami cieczy. Jednakże w przypadku dyspergowania przy elektrorozpylaniu, ciecz wypływa z kapilary nie tylko pod wpływem pola elektrycznego, lecz także pod wpływem czynnika mechanicznego[1].
cząsteczkami posiadającego wysoką temperaturę gazu otaczającego, co powoduje odparowywanie rozpuszczalnika z kropel pod wpływem temperatury, oraz na skutek mechanicznych zderzeń naładowanych kropel z cząsteczkami gazu[11,17]. Ponieważ cząsteczki rozpuszczalnika odparowują z
naładowanych kropli z szybkością o wiele wyższą od szybkości odparowywania jonów analitu[2], rozmiar kropli ciągle się zmniejsza przy
jednoczesnym zachowaniu wielkości ładunku. To powoduje znaczny wzrost stężenia jonów analitu podczas zmniejszania kropli, a więc powierzchniowa gęstość ładunku kropli wzrasta[1,2,11]. Proces ten ma miejsce do osiągnięcia przez kroplę tak zwanej granicy stabilności Rayleigha (ang. Rayleigh
stability limit, RSL), przy której siła elektrostatycznego odpychania
równoimiennych ładunków staje się równa sile napięcia powierzchniowego cieczy. Blisko osiągnięcia granicy Rayleigha, dochodzi do rozpadu kropli z utworzeniem mniejszych kropli wtórnych – zjawiska, znanego jako rozpad kulombowski (ang. Coulomb fission)[3,5,15]. Rozpad kulombowski może
zostać opisany za pośrednictwem aparatu matematycznego teorii katastrof (ang. the catastrophe theory)[18], gdyż ciągła zmiana jednego z parametrów
układu (a mianowicie gęstości powierzchniowej ładunku) doprowadza do nieciągłej zmiany stanu układu – utraty stabilności przez stabilną poprzednio kroplę i gwałtownego przejścia do stabilnego w nowych warunkach stanu, właściwego dla kropli wtórnych. Krople wtórne ulegają dalszym rozpadom kulombowskim, co prowadzi do utworzenia kropel o wymiarach rzędu kilku nanometrów i o ładunku rzędu kilku ładunków elementarnych[5]. Przyczyną
rozpadu kulombowskiego kropli poniżej granicy Rayleigha są deformacje kształtu kropli[2,9]. Powierzchnia naładowanej nanokropli w praktyce nie jest
ani sferyczna, ani gładka. Na skutek obecności ładunku na powierzchni kropli, znajdującej się w polu elektrycznym, następuje wzmocnienie falowania jej powierzchni[2]. Dla silnie naładowanych kropli deformacje te
powierzchni kropli mogą wystąpić wyraźne wypukłości, które z kolei ułatwiają oddzielenie kropli wtórnych: najpierw pojedyńczy jon wywołuje wzrost wypukłości, a następnie poprzez odpychanie elektrostatyczne niektóre jony, znajdujące się w wewnętrznej części kropli, są wypychane do tej wypukłości. Gdy odpychanie pomiędzy resztą kropli a oddzielonymi w ten sposób jonami przewyższy siły kohezyjne w środku wypukłości, oddziela się kropla wtórna[2,15]. Wzajemna polaryzacja sąsiadujących kropli także może prowadzić do deformacji i powodować rozpad kropli poniżej granicy Rayleigha[2]. Oprócz tego, deformacja kropli w polu elektrycznym może także polegać na przyjęciu kształtu wydłużonego lub spłaszczonego[9,11,16]. Podobna zmiana symetrii kropli może być przyczyną
złożonych ruchów mechanicznych, które również mogą powodować rozpad kropli na mniejsze części[16].
Utworzenie jonów w fazie gazowej
Chociaż ESIMS jest obecnie jedną z najszerzej stosowanych w chemii, biologii, medycynie i biotechnologii technik spektrometrii mas, ciągle prowadzone są dyskusje dotyczące mechanizmu tworzenia jonów w fazie gazowej[1]. Istnieją dwa mechanizmy tworzenia jonów w fazie gazowej z bardzo małych, wysoce naładowanych kropli, uznawane przez licznych autorów za najważniejsze[5,11,13,19-21], a opracowane przez Dole’a i wsp.[22] oraz Iribarne’a i Thomsona[23,24].
Mechanizm Dole’a[5,11,19-21] [rys. 2, (a)] polega na tym, że rozpady
kulombowskie (RK) kropli (w tym kropli wtórnych) prowadzą ostatecznie do utworzenia kropli, zawierającej tylko jedną cząsteczkę analitu. Po odparowaniu całego rozpuszczalnika, ładunek kropli pozostaje na tej pojedynczej cząsteczce analitu, i w ten sposób zostaje utworzony „wolny” jon w fazie gazowej. Istotą mechanizmu Dole’a są więc rozpady
kulombowskie. Po odparowaniu ostatnich cząsteczek rozpuszczalnika, nadmiar ładunku lokalizuje się w miejscu (lub miejscach, jeśli cząsteczka analitu posiada kilka możliwych miejsc wiązania ładunku), które zapewnia utworzenie najbardziej stabilnego jonu analitu w fazie gazowej[11].
Mechanizm ten jest znany pod nazwą mechanizmu pozostałości ładunku lub mechanizmu naładowanej reszty (ang. charge residue model[5,13,25] lub
charged residue model[11,13,19,25,26], CRM).
s s s s s + s s s s s s s s + + + + + + + + +s s s s ++ +s s+ ss s + + + + + s s s s s s s s++ + s s s s s s s s s s s + s s s s s s s s + + + + + + + +s s s ++ +s s+ s + + + + + s s s + s + s s s s s s + + + + + + s s+ + +s s s + + +++ a) b) RK RK EJ c) d)
Rys. 2. Utworzenie jonów w fazie gazowej (tryb jonów dodatnich, przy założeniu, że roztwór składa się z całkowicie zjonizowanych cząsteczek analitu, a rozdzielenie ładunków dodatnich i ujemnych w kapilarze zaszło w 100%): a) – według mechanizmu Dole’a, b) – według mechanizmu Iribarne’a i Thomsona, c) – odparowywanie rozpuszczalnika w mechaniźmie CRM, d) – odparowywanie jonu w mechaniźmie IEM; s - cząsteczki rozpuszczalnika, + – kationy, RK – rozpad kulombowski, EJ – emisja jonu.
Mechanizm Iribarne’a i Thomsona[11,19-21,25] [rys. 2, (b)] polega na
zmniejsza się do rozmiaru rzędu 10-20 nm [13], przy czym pole elektryczne
uwarunkowane gęstością ładunku na powierzchni kropli staje się na tyle duże (rzędu 109 V·m-1)[20,24], że następuje nie rozpad kropli na mniejsze
krople, lecz emisja solwatowanych jonów (EJ na rys. 2) bezpośrednio do fazy gazowej. Odparowywanie jonu zastępuje więc rozpad kulombowski[13,26]. Proces ten jest podobny do procesu oderwania kropel
wtórnych od ulegających deformacjom kropel macierzystych: emitowany jon doznaje działania dwóch konkurujących sił elektrostatycznych – siły przyciągania do cząsteczek rozpuszczalnika i siły odpychania od równoimiennych jonów znajdujących się na powierzchni kropli[23-27]. To
spowodowane jest obecnością bariery energetycznej, znajdującej się w pewnej odległości na zewnątrz od powierzchni kropli[13,20,23,27]. Dla układu
kropla-jon uwolnienie jonu w postaci solwatowanej jest w tym przypadku korzystniejsze energetycznie[26]. Ważną rolę w tym mechanizmie odgrywa
zdolność jonów do przemieszczania się w kierunku powierzchni kropli. Istotą tego mechanizmu jest odpychanie między jonami na powierzchni kropli a resztą jonów w jej wnętrzu[12]. W literaturze ten mechanizm jest
znany jako mechanizm desorpcji (odparowywania) jonu (ang. ion desorption
model, IDM[5], ion evaporation model, IEM[11,26], lub field emission[24,25]).
Zasadnicza różnica pomiędzy mechanizmami Dole’a a Iribarne’a i Thomsona polega na sposobie oddzielenia jonu analitu od reszty kropli[5].
Według CRM oddzielenie zachodzi na skutek odparowywania rozpuszczalnika [rys. 2, (c)] szeregu rozpadów kulombowskich, które ostatecznie doprowadzają do powstania kropli, zawierającej tylko jedną cząsteczkę analitu. Natomiast według IEM oddzielenie zachodzi, gdy pojedynczy solwatowany jon analitu zostaje desorbowany do fazy gazowej [rys. 2, (d)], osłabiając w ten sposób odpychanie kulombowskie pomiędzy jonami w kropli. W mechanizmie CRM przyjmuje się, że przez powierzchnię rozdziału faz przechodzą cząsteczki rozpuszczalnika,
natomiast w IEM – jony [rys. 2, (c), (d)]. Mechanizm tworzenia jonu w fazie gazowej jest ściśle związany z wartością energii swobodnej przeniesienia jonu z powierzchni naładowanej kropli do fazy gazowej (∆G◦) – im mniejsza
jest ta energia, tym większe jest prawdopodobieństwo tworzenia jonów w fazie gazowej poprzez mechanizm IEM[26]. Jednak różnica między rozpatrywanymi mechanizmami tworzenia jonów w fazie gazowej może być niewyraźna, jeśli porównuje się desorpcję pojedyńczego solwatowanego jonu z kropli (IEM) z ostatnim z szeregu kulombowskich rozpadów, po którym zostaje kropla potomna zawierająca tylko jedną solwatowaną cząsteczkę analitu i nadmiar ładunku (CRM). Poza tym, nie ma żadnej wyraźnej różnicy w stopniu naładowania lub solwatacji jonów, utworzonych według tych dwóch mechanizmów[11].
Trudno ocenić, który z tych dwóch mechanizmów bardziej odpowiada rzeczywistym procesom zachodzącym podczas tworzenia jonów w fazie gazowej, gdyż modele te zostały oparte na obserwacji cząsteczek o zupełnie odmiennych właściwościach fizykochemicznych, przede wszystkim o dalekich od siebie masach: model Dole’a powstał w oparciu o badania zachowania się makrocząsteczek (makrojonów frakcji polistyrenu o masach 51000 Da i 411000 Da)[22], podczas gdy model Iribarne’a i Thomsona
powstał w oparciu o badania małych jonów (proton, kationy metali alkalicznych, kationy tetraalkiloamoniowe oraz bardziej lub mniej złożone aniony organiczne i nieorganiczne)[23,24]. Trudne jest zatem przełożenie tych mechanizmów na cząsteczki o innych, nieuwzględnionych przez autorów rozmiarach[13]. Najbardziej prawdopodobne jest to, że proces tworzenia
jonów w fazie gazowej przebiega według obu tych mechanizmów w zależności od natury chemicznej analitu. Natomiast żaden z tych mechanizmów nie może w pełni uzasadnić wszystkich obserwacji[5].
Przyjęto, że formy jonowe dużych cząsteczek (o masach powyżej 3300 Da)[2,11,13,27], w tym jonów wielokrotnie naładowanych[2], w ESMS
tworzone są według mechanizmu CRM. Spór nadal dotyczy mechanizmu desorpcji mniejszych jonów, ale większość autorów skłania się raczej ku mechanizmowi IEM[11,13,19,21,28]. Nie ma natomiast eksperymentalnych
dowodów na korzyść któregoś z tych dwóch mechanizmów, zwłaszcza na poziomie nanokropli, gdyż do wyjaśnienia różnic, trzeba by było całkowicie odizolować te dwie drogi tworzenia jonów w fazie gazowej, co jest bardzo trudne technicznie.
2. Czynniki wpływające na kształt widma mas
W elektrorozpylaniu, będącym głównie sposobem przeniesienia z roztworu do fazy gazowej jonów już istniejących w roztworze[5,12] bardzo
ważną rolę odgrywa etap desolwatacji jonów w fazie gazowej, mający swoje odzwierciedlenie w widmie mas. Kształt widma mas zależy od czynników, wpływających na proces uwolnienia jonu od otoczki solwatacyjnej, a także od stężenia analitu i towarzyszących elektrolitów oraz od reakcji elektrochemicznych, zachodzących głównie w kapilarze.
3.1. Czynniki wpływające na proces desolwatacji jonów w fazie gazowej a) Wpływ wartości napięcia przykładanego do kapilary na proces
desolwatacji jonów w fazie gazowej
Oddziaływanie przykładanego do kapilary pola elektrycznego na roztwór analitu jest początkowym etapem procesu elektrorozpylania. W granicach zwykle stosowanego napięcia na kapilarze (2 – 10 kV) można wyróżnić dwa charakterystyczne przedziały, różniące się wielkością napięcia przykładanego do kapilary: przedział niskiego (ang. low-voltage mode) oraz wysokiego napięcia (ang. high-voltage mode)[10].
Przy przykładaniu do kapilary niskiego napięcia uzyskuje się sprej o kształcie pojedynczego stożka, o podstawie przyległej do brzegu
kapilary[10,21,28]. Obserwowane w widmie mas sygnały odpowiadają jonom
składającym się z jonu analitu, zagregowanego lub nie z jedną lub kilkoma niezjonizowanymi cząsteczkami analitu lub rozpuszczalnika[21].
Przy przykładaniu do kapilary wysokiego napięcia następuje przejście do warunków, w których z licznych miejsc, ulokowanych dookoła brzegu kapilary, tworzy się subtelna mgiełka kropli, a centralna struga cieczy zanika[23, 25]. Jednocześnie silnie wzrasta całkowity prąd jonowy[21]. Yamashita i Fenn tłumaczą te zmiany początkiem wyładowywania koronowego2 oraz zapoczątkowaniem reakcji jonowo-molekularnych, które to wyładowywanie może spowodować. Wartość potencjału (5 kV [21] lub
9 kV [28]), charakterystyczna dla granicy pomiędzy przedziałem niskiego
napięcia a przedziałem wysokiego napięcia, zależy przeważnie od natężenia pola elektrycznego, a także w pewnym stopniu od składu cieczy, formy i rodzaju materiału wykończenia końcówki kapilary, jak również prawdopodobnie od ciśnienia i składu otaczającego gazu[10,21]. W tym
przedziale potencjału dochodzi do desolwatacji jonów oraz do reakcji chemicznych – fragmentacji[10] i różnorodnych reakcji
jonowo-molekularnych[21], co znajduje swoje odzwierciedlenie na widmie mas: zawiera ono sygnały pochodzące od niesolwatowanych jonów analitu, a także od jonów fragmentacyjnych oraz szumy[21].
Zmiany charakteru spreju, stopnia desolwatacji jonów w fazie gazowej oraz widma mas w zależności od wartości napięcia przykładanego do kapilary obserwowane są zarówno w trybie jonów dodatnich, jak i ujemnych. Natomiast znak potencjału przykładanego do kapilary wpływa na natężenie prądu jonowego oraz wartość napięcia, przy którym dochodzi do wyładowywania: prądy jonowe jonów ujemnych są nieco niższe od prądu
2 Wyładowywanie koronowe – wyładowywanie elektryczne w gazie przy ciśnieniu rzędu 1 atm, w silnie niejednorodnym polu elektrycznym; występuje na elektrodach o małym promieniu krzywizny[4].
jonów dodatnich, uzyskanych w analogicznych warunkach, a z kolei napięcie na kapilarze, przy którym dochodzi do wyładowywania koronowego w przypadku jonów ujemnych także jest nieco niższe od tego dla jonów dodatnich[21].
b) Wpływ rozpuszczalnika i gazu kolizyjnego na proces desolwatacji jonów w fazie gazowej
W ESMS stosowane są rozpuszczalniki (lub mieszaniny rozpuszczalników), łączące właściwości wysokiej lotności, wysokiej polarności oraz zdolności rozpuszczania wielu różnorodnych związków. Są to przede wszystkim: woda[13,29], metanol[13,30,31], propanol[27], acetonitryl[13,32]
lub ich mieszaniny[33-36]. Rzadziej używa się formamidu[27,13], acetonu[22] i
benzenu[22]. Lotne rozpuszczalniki pozwalają na szybkie odparowywanie
kropli, a więc na skuteczność tworzenia jonów w fazie gazowej[10,21].
Rozpuszczalniki polarne obniżają wielkość potencjału elektrycznego, niezbędnego do rozpoczęcia rozpylania elektrostatycznego cieczy[9,13] oraz
zwiększają prąd w kapilarze[11]. Sprzyjają one również rozdzieleniu
ładunków w roztworze[11,13], w tym elektroforetycznemu rozdzieleniu kationów i anionów w kapilarze, co niesie następujące skutki: (i) utrudnianie przeniesienia przeciwjonów do kropli wtórnych lub zmniejszenie skłonności przeciwjonów do pozostania przyłączonymi do wielokrotnie naładowanych jonów analitu w fazie gazowej[11,13]; (ii) tworzenie raczej jonu [M – H]¯, niż np. [M + Cl]¯, gdyż właśnie ten pierwszy jest produktem reakcji rozdzielenia ładunku[11]; (iii) tworzenie jonów
naładowanych wielokrotnie[13], poprzez mechanizm reakcji dysocjacji[11]:
AX
2AX
++ X
-Proces desolwatacji jonów jest procesem endotermicznym i energia potrzebna do jego realizacji w ESIMS jest dostarczana do kropli poprzez zderzenia z cząsteczkami gazu o wysokiej temperaturze[1,20]. Wiodącą rolę
gazu kolizyjnego w procesie tworzenia jonów w fazie gazowej potwierdza porównanie elektrorozpylania z techniką elektrohydrodynamicznej spektrometrii mas (ang. electrohydrodynamic mass spectrometry, EHMS)[1,19], polegającą na dyspergowaniu do próżni roztworu elektrolitu rozpuszczonego w nisko lotnym rozpuszczalniku. W przypadku EHMS, odparowywanie rozpuszczalnika zachodzi wyłącznie pod wpływem oddziaływania pola elektrycznego na ciecz przy końcówce kapilary i jest mało efektywne w porównaniu z odparowywaniem rozpuszczalnika mającym miejsce przy ESIMS, gdzie odparowywanie rozpuszczalnika zachodzi także na skutek zderzeń z cząsteczkami gazu otaczającego. Poza tym, otaczający gaz w ESIMS służy także do zmniejszenia energii kinetycznej naładowanych kropli oraz jonów z nich powstających. Brak gazu otaczającego (EHMS) powodowałby, że powstające jony miałyby zbyt dużą energię kinetyczną w stosunku do wymagań najczęściej stosowanych analizatorów mas[1,10]. Zwiększenie ciśnienia gazu otaczającego w ESMS powoduje pełniejszą desolwatację jonów[17].
Ponadto gaz przepływający utrzymuje wnętrze analizatora mas w czystości oraz wspomaga dysocjację klasterów jonów analitu[10,28,37]. Dla
zapobieżenia kondensacji i klasteryzacji jonów istotne jest, żeby gaz ten był wolny od pary rozpuszczalnika, co osiąga się przez stosowanie wysokiej szybkości przepływu gazu, dzięki czemu para rozpuszczalnika jest nieprzerwanie wydalana z komory jonizacyjnej.
3.2. Wpływ stężenia analitu i towarzyszących elektrolitów na kształt widma mas
Obserwować na widmie mas sygnały, pochodzące od jonów analitu, można tylko w pewnym przedziale jego stężeń – poniżej 10-2 M [28].
Związane jest to z efektywnością rozdzielenia ładunków dodatnich i ujemnych w obszarze stożka Taylora, zachodzącego pod wpływem pola elektrycznego przykładanego do kapilary. Przy niskich stężeniach elektrolitu (poniżej 3×10-5 M) następuje efektywne, bo blisko 40%-owe, rozdzielenie
ładunków, przy stężeniach rzędu 10-3 M rozdział ten jest prawie dziesięciokrotnie gorszy, natomiast przy stężeniach elektrolitu rzędu 10-2 M
rozdzielenie ładunków zachodzi tylko w 0.3% przypadków[13,28]. W związku
z tym przy stężeniach powyżej 10-5 M czułość względem analitu spada wraz
ze wzrostem stężenia analitu i obecnością obcego elektrolitu w roztworze. Ten spadek efektywności rozdzielenia ładunku3 prowadzi w konsekwencji
do odpowiedniego spadku efektywności detekcji analitu za pomocą spektrometru mas i do spadku intensywności sygnału, pochodzącego od jonu analitu[28].
Elektrolity towarzyszące odgrywają podwójną rolę w kształtowaniu widma mas. Z jednej strony, elektrolity towarzyszące w sposób nieunikniony z różnych źródeł trafiają do badanej próbki: z rozpuszczalnika (np. woda i metanol mogą zawierać do ~10-5 M Na+)[13,20], z buforu, z zanieczyszczeń
kapilary lub części aparaturowych. Poza tym elektrolity są często celowo dodawane do roztworu próbki badanej w celu wspomagania tworzenia specyficznych typów jonów (np. kwasy są dodawane celem otrzymania jonów MH+, zasady – jonów [M – H]–); sole dodawane są celem otrzymania
3 Efektywność rozdzielenia ładunku można zwiększyć, zmniejszając szybkość przepływu: przy nadzwyczaj niskich szybkościach przepływu (w tzw. trybie „nano-przepływu”, 10 – 100 nl·min-1), efektywność rozdziału jonów dodatnich i ujemnych w obszarze stożka Taylora może osiągnąć nawet 100% [11].
jonowych form związków, nie posiadających grup funkcyjnych, uczestniczących w równowadze kwasowo-zasadowej[14,20,34,36,38]. W tym
przypadku mówi się o pomocniczej funkcji towarzyszącego elektrolitu, gdyż jego jony kumulują się na powierzchni kropli tak samo jak jony analitu i przyczyniają się w ten sposób do wzrostu gęstości ładunku na powierzchni kropli oraz do osiągnięcia przez kroplę stanu umożliwiającego emisję jonu według IEM[11,14,27]. Według Bladesa i wsp.[14] minimalne stężenie elektrolitu w badanej próbie musi być takie, by roztwór wykazywał przewodnictwo σ blisko 3×10-7 Ω-1·cm-1, natomiast przy σ = 1×10-7 Ω-1·cm-1 elektrorozpylanie traci stabilność[14].
Z drugiej strony, zbyt duże stężenie towarzyszącego elektrolitu może wywołać spadek intensywności sygnału analitu, które może uniemożliwić analizę złożonych mieszanin, zwłaszcza przy śladowych ilościach interesującej substancji. Zjawisko to jest jedną z głównych wad elektrorozpylania jako techniki stosowanej w spektrometrii mas i wymaga przeprowadzenia uprzedniego rozdzielenia mieszanin, np. za pomocą chromatografii[13]. Spadek intensywności sygnału analitu jest uwarunkowany
tym, że towarzyszący elektrolit jako składowa część naładowanych kropli równo z analitem uczestniczy w tworzeniu w kropli powierzchniowej warstwy jonowej i konkuruje z jonami analitu w desorpcji z powierzchni kropli[13,28]. Towarzyszący elektrolit może także poprzez efekty jonowe
zmniejszyć stopień rozdziału dodatnich i ujemnych jonów analitu w obszarze stożka Taylora[28]. Ponieważ efektywność rozdzielenia ładunków
dodatnich i ujemnych zmniejsza się wraz ze wzrostem ogólnego stężenia elektrolitu, a emisja jonów z kropli jest procesem nieselektywnym, czułość tej techniki na jony analitu zmniejsza się nie tylko ze wzrostem stężenia analitu (j.w.), lecz także ze wzrostem całkowitego stężenia elektrolitu w rozpraszanym elektrostatycznie roztworze[28]. Spadek intensywności sygnału
zawartych w roztworze[13]. Ilość jonów przeniesionych z roztworu do fazy
gazowej zależy od ich stężenia w roztworze oraz od współczynnika, który wyraża względną efektywność przeniesienia jonów do fazy gazowej i którego wielkość zależy od mechanizmu tworzenia jonów w fazie gazowej. Z tego powodu jony powierzchniowo czynne, mające wysoką wartość tego współczynnika i wykazujące tendencję do kumulowania się na powierzchni kropli są niepożądane ze względu na powodowanie spadku stężenia jonów analitu na powierzchni kropli i bardzo intensywne sygnały w widmie mas, interferujące z widmem mas analitu[13].
3.3. Wpływ reakcji elektrochemicznych, zachodzących w kapilarze, na kształt widma mas
Reakcje elektrochemiczne mogą wpływać na kształt widma mas. Np. przy pracy z kapilarą stalową lub cynkową w widmach mas próbek nie zawierających w swoim składzie kationów żelaza lub cynku, w odpowiednich warunkach można zaobserwować sygnały pochodzące od jonów zawierających kationy tych metali[14]. Dzieje się tak z tego powodu,
że na granicy faz metal-roztwór w kapilarze dochodzi do (i) przekształcenia cząsteczek obojętnych w jony oraz do (ii) przekształcenia przeciwjonów w cząsteczki obojętne[1,11,13]. Obecność przynajmniej jednego z tych dwóch procesów jest niezbędna do podtrzymywania ciągłości procesu tworzenia naładowanych kropli[1]. Reakcje elektrochemiczne są zatem częścią składową rozdzielenia ładunków w obszarze stożka Taylora. Zmiany składu roztworu zachodzące wskutek procesów elektrochemicznych w dużym stopniu zależą od wielkości napięcia przykładanego do kapilary, szybkości przepływu (przy niskich szybkościach przepływu i wysokich wartościach napięcia przykładanego do kapilary procesy elektrochemiczne zachodzą bardziej ekstensywnie[1]) i stężenia roztworu oraz od właściwości
może zależeć nie tylko od roztworu, lecz także od materiału konstrukcyjnego i stanu kapilary[1,13]. Np. kationy srebra, miedzi i rtęci przy pracy
spektrometru w trybie jonów ujemnych mogą ulegać elektrolitycznej redukcji i osadzać się na częściach konstrukcyjnych źródła jonów, zmieniając ich właściwości powierzchniowe, w tym właściwości oksydacyjno-redukcyjne[1,39]. Z kolei reakcje utleniania żelaza i cynku,
zawartych w materiale konstrukcyjnym kapilary prowadzą do jej elektrochemicznego rozpuszczania[14].
Źródło elektrorozpylania działa zatem jako pewnego rodzaju ogniwo elektrochemiczne[39,14,13]. Dzięki powstawaniu jonów wskutek reakcji
elektrochemicznych, elektrorozpylanie staje się techniką jonizacji, a nie tylko techniką przeniesienia jonów z roztworu do fazy gazowej[12,13].
Przy zastosowaniu roztworów wodnych, na kształt widma mas mogą wpłynąć także zmiany pH, spowodowane reakcjami elektrochemicznymi wody[27]. W tych przypadkach podczas procesu elektrorozpylania zmienia się
stosunek stężeń protonów do stężeń innych kationów w roztworze, co może wpływać na intensywności pików pochodzących od cząsteczek protonowanych w porównaniu z intensywnością pików pochodzących od innych kationów w roztworze; poza tym, wywiera to nieznaczny wpływ na stopień naładowania cząsteczek wielokrotnie naładowanych. Czynniki te mogą mieć istotne konsekwencje dla analizy ilościowej[27].
3. Intensywność sygnałów w widmie mas
Intensywność sygnałów, pochodzących od poszczególnych jonów w widmie mas zależy od właściwości roztworu: stężenia, reaktywności oraz właściwości fizykochemicznych jego składników, charakteru i siły oddziaływań pomiędzy jonami analitu i rozpuszczalnika oraz skuteczności desolwatacji jonów[10,17,21,29,38].
Oprócz tego, intensywność sygnału jonu w widmie mas zależy od warunków doświadczalnych (np. szybkości przepływu roztworu badanego przez kapilarę, rozwiązania konstrukcyjnego źródła jonów), które wpływają na to, jaka część jonów, zawartych w kropli, jest przetwarzana na jony w fazie gazowej oraz od tego, na ile w spektrometrze mas skuteczna jest detekcja jonów w porównaniu z ogólną ilością tych samych jonów, przeniesioną do fazy gazowej[13].
Efektywność przeniesienia jonu z naładowanej kropli do fazy gazowej jest ściśle związana z mechanizmem tworzenia jonu w fazie gazowej. Proste jony nieorganiczne dzięki łatwości oderwania się od naładowanej kropli, oraz związki hydrofobowe i powierzchniowo czynne dzięki zdolności kumulacji na powierzchni kropli są podatne na uwolnienie z kropli poprzez mechanizm IEM[13,26]. Natomiast związki hydrofilowe, jak
np. proteiny globularne, nie mające tendencji do przemieszczania się w kierunku powierzchni kropli, podlegają przeniesieniu do fazy gazowej za pomocą mechanizmu CRM[13].
Efektywności przeniesienia jonów z naładowanych kropli do fazy gazowej są podobne dla podobnych pojedyńczo naładowanych jonów (jak np. kationy metali alkalicznych). Z kolei dla jonów, które kumulują się na powierzchni (jony związków powierzchniowo czynnych, jony związków hydrofobowych), efektywność ta wzrasta wraz ze wzrostem właściwości hydrofobowych tych jonów[13,26]. Np. dla (C
2H5)4N+ efektywność
niż dla Cs+, (n-C
3H7)4N+ – 5 razy większa, (n-C7H15)NH3+ – 8 razy większa,
(n-C5H11)4N+ – 16 razy większa[13]. Jednakże związki, mające w swojej
budowie bardzo długie łańcuchy alkilowe (np. kwas stearynowy, alkohol cetylowy, alkohol decylowy), są tak bardzo hydrofobowe, że tworzą na powierzchni kropli film, zapobiegający odparowywaniu rozpuszczalnika i w ten sposób tworzeniu jonów w fazie gazowej[26].
Na jakościowy kształt widma mas i intensywność obserwowanych sygnałów mogą wpłynąć reakcje, mające miejsce w fazie gazowej, jak na przykład reakcje przeniesienia protonu. Składnik roztworu (w tym także rozpuszczalnik), który ma wyższą zasadowość w fazie gazowej, może poprzez reakcję przeniesienia protonu w fazie gazowej zmniejszyć intensywność sygnału jonu analitu o niższej zasadowości w fazie gazowej, a niekiedy całkowicie uniemożliwić jego obserwację[13].
4. Stan energetyczny jonów tworzonych podczas elekrorozpylania Jony tworzone podczas elektrorozpylania mają bardzo mały nadmiar energii wewnętrznej[11,38], dlatego elektrorozpylanie nazywane jest
najbardziej „miękkim” ze wszystkich obecnie znanych sposobów tworzenia jonów w fazie gazowej[11,38,40,19]. Wprawdzie na roztwór badanej substancji
podczas jego przepływu przez kapilarę działa bardzo silne pole elektryczne, jednak energia tego pola nie jest przekładana bezpośrednio na jony, tylko na roztwór, i jest zużywana na szereg następujących po sobie procesów tworzenia jonów w fazie gazowej. Większość energii wewnętrznej (jest to przede wszystkim energia drgań oscylacyjnych i rotacyjnych[11])
solwatowanych jonów jest rozpraszana podczas desolwatacji4. Wobec tego
4 Np. w najstarszej ze stosowanych w spektrometrii mas technik jonizacji – jonizacji poprzez bombardowanie strumieniem elektronów (ang. electron ionization, electron impact, EI), strumień elektronów o energii kilkudziesięciu elektronowoltów oddziaływuje na parę badanej substancji. Wobec tego energia wewnętrzna, nabyta przez cząsteczkę analitu podczas zderzenia z elektronem, może być przekazywana
dla procesu elektrorozpylania charakterystyczny jest bardzo niski stopień fragmentacji[11,19,38]. W tej postaci ESMS jest stosowana do określenia mas
cząsteczkowych związków i badania składu mieszanin[5,19,41-48], obserwacji
oddziaływań międzycząsteczkowych[38], w tym procesów samoorganizacji w
roztworach zasad nukleinowych[36], oddziaływań niekowalencyjnych pomiędzy cząsteczkami białek i niektórych leków (np. o budowie porfirynowej) z DNA oraz RNA[49-51].
Jeśli po całkowitym odparowaniu rozpuszczalnika „wolny” jon w fazie gazowej wciąż posiada nadmiar energii wewnętrznej, możliwe stają się trzy procesy: utlenianie, redukcja lub fragmentacja jonu[52]. Ten ostatni
proces pozwala na uzyskanie cennych informacji dotyczących struktury badanego związku[10,19,37,53-55]. Badania strukturalne za pomocą ESMS są
możliwe dzięki realizacji przez jon randomizacji[19,37,53] energii wewnętrznej
względem wszystkich oscylacyjnych stopni swobody w ciągu kilku pikosekund[53] bezpośrednio przed reakcją fragmentacji; w wyniku tej
randomizacji, ze wszystkich możliwych dróg rozpadu jon wybiera tę termodynamicznie najbardziej korzystną[19,37], a widmo mas nie jest
nieuporządkowane, lecz zawiera sygnały pochodzące od jonów, utworzonych na skutek rozerwania najsłabszych wiązań w cząsteczce. Dodatkowym potwierdzeniem uporządkowania procesu fragmentacji jonu jest ten fakt, że jony, utworzone wskutek różnych metod jonizacji ulegają rozpadowi według tych samych dróg fragmentacyjnych; różnica polega tylko na stopniu rozpadu, gdyż zależy on od ilości energii, przekazywanej jonowi podczas jonizacji[56].
Wymuszenie reakcji fragmentacji jonów w celu uzyskania informacji strukturalnych znalazło swoje zastosowanie w technice, znanej jako dysocjacja zderzeniowa lub fragmentacja aktywowana/indukowana tylko wewnątrz utworzonego jonu, w wyniku czego najczęściej zachodzi jego
kolizją (ang. collisionally activated dissociation, CAD[19,38], lub collisionally
induced dissociation, CID[19,38]). Technika ta polega na tym, że stabilny jon,
posiadając energię kinetyczną, ulega kontrolowanym kolizjom z cząsteczkami lub atomami chemicznie obojętnego gazu (N2[31,57], Ar[19,57],
He[19,53], H2[19,53], Kr[57], a także CH4[57] lub C3H8[57]). Na skutek tych kolizji,
część energii kinetycznej jonu przekształca się w jego energię wzbudzenia elektronowego, która z kolei przekształca się w energię wzbudzenia wibracyjnego i ulega randomizacji[53,57]. Do opisania tego mechanizmu
przekazywania energii jonu podczas CID można zastosować teorię quasi-równowagi (ang. quasi-equilibrium theory, QET)[19,53,55]. Według tej teorii,
maksymalna energia wewnętrzna, którą może nabyć jon podczas jednego zderzenia, określana jest jako energia środka masy[37]:
(
0 str)
2 1 2 śmE
q
V
E
m
m
m
E
+
∆
−
+
=
, (1) gdzie Eśm – energia środka masy, m2 – masa cząsteczkowa gazu kolizyjnego,m1 - masa jonu, E◦ – początkowa energia wewnętrzna jonu, q – ładunek jonu,
∆V - różnica potencjałów, wyznaczająca energię kinetyczną jonu, Estr - energia kinetyczna stracona przez jon podczas kolizji. Zgodnie z równ.
(1), odpowiednio wysoka energia środka mas może być przekazana jonowi poprzez zastosowanie cięższego gazu kolizyjnego lub poprzez nadanie jonowi większej energii kinetycznej. Jeśli energia kinetyczna jonu ulegającego kolizji ma wartość od kilku do kilkudziesięciu keV, mówi się o kolizjach wysokoenergetycznych (ang. high-energy collisions)[19,37] i stosuje się lżejszy gaz kolizyjny (np. He)[19]. Natomiast gdy energia kolizyjna
wynosi 1 – 200 eV, mówi się o kolizjach niskoenergetycznych (ang.
low-collision energy)[19,37] i stosuje się cięższy gaz kolizyjny (np. N
2, Ar, Xe)[19].
Zwiększenie energii wewnętrznej jonu osiąga się także poprzez zwiększenie intensywna fragmentacja[19,38].
ilości zderzeń, przy czym kilkaset niskoenergetycznych kolizji i kilka (9 - 10) zderzeń wysokoenergetycznych powodują jednakowe zwiększenie wartości energii wewnętrznej jonu[37], co pozwala na zmianę parametrów
doświadczalnych w zależności od posiadanego wyposażenia bez istotnych zmian w charakterze fragmentacji.
Parametry doświadczalne oraz różnice projektowe źródła jonów wywierają znaczny wpływ na wartość energii wewnętrznej jonów[11]: zależy ona przede wszystkim od techniki jonizacji5. Przy wzroście masy
cząsteczkowej substancji przeprowadzenie badań strukturalnych nad jonami naładowanymi pojedyńczo za pomocą CID staje się trudne ze względu na niemożliwość wzbudzenia kolizyjnego takich jonów, uwarunkowaną małą wartością energii środka masy. W przypadku jonu naładowanego wielokrotnie, energia ta zwiększa się [zwiększa się składnik q∆V równ. (1)], co umożliwia przeprowadzenie badania struktury substancji za pomocą CID[34].
Jeśli nabyta w ten sposób przez jon energia przewyższa energię któregoś z wiązań, obecnych w jonie, zachodzi jonowo-molekularna reakcja rozpadu wzdłuż tego wiązania i na widmie mas obserwuje się sygnał(-y), pochodzący(-e) od odpowiedniego(-ich) jonu(-ów)[53]. Na określenie
struktury jonu pozwala analiza m/z sygnałów, pochodzących od jonów fragmentacyjnych, oraz stosunku intensywności różnych sygnałów obecnych na widmie mas[53]. Badania stosunku intensywności sygnałów pochodzących od jonów fragmentacyjnych, powstałych wskutek CID, jako funkcji różnych parametrów pomiarowych przeprowadził McLafferty[53-55]. Odkrył on, że
przy zwiększeniu wartości energii kinetycznej jonu, ulegającego zderzeniu,
5 W EI jon uzyskuje duży nadmiar energii wewnętrznej, który powoduje intensywną fragmentację analitu, natomiast w ES większość nabytej przez roztwór analitu w kapilarze energii jest rozpraszana podczas desolwatacji. Z kolei w desorpcji jonu wspomaganej matrycą, energia wewnętrzna jonu bardzo zależy od rodzaju stosowanej matrycy[58].
nie tylko zwiększa się ilość rodzajów i liczebność jonów fragmentacyjnych, lecz także zmienia się stosunek intensywności sygnałów, pochodzących od tych fragmentów. Przy wyższych wartościach energii kinetycznej jonu oraz ciśnienia gazu kolizyjnego obserwuje się wzmocnienie procesu fragmentacji o większej energii aktywacji. Na widmie mas odzwierciedla się to większym wzrostem intensywności sygnałów pochodzących od jonów fragmentacyjnych, tworzonych w reakcji o większej energii aktywacji, niż tych, tworzonych w reakcjach o mniejszej energii aktywacji[53].
Według QET, stosunek intensywności sygnałów, pochodzących od jonów fragmentacyjnych, zależy od rozkładu energii w jonie macierzystym bezpośrednio przed zderzeniami oraz od stałych szybkości poszczególnych reakcji rozpadu[53]. Z kolei stałe szybkości reakcji rozpadu jonu są funkcją
częstotliwości oscylacyjnych i rotacyjnych, a także energii aktywacji reakcji[53]. To pozwala na rozróżnienie dzięki CID jonów izomerycznych,
gdyż mają one różne częstotliwości wibracyjne i rotacyjne oraz różne energie aktywacji odpowiednich reakcji fragmentacji[53]. Reakcja
izomeryzacji może zastępować[53], uprzedzać lub zachodzić tuż po[52] reakcji
fragmentacji w zależności od bariery energetycznej tych dwóch reakcji[19,53]. Reakcje fragmentacji, przeprowadzane za pomocą spektrometrii mas pozwoliły na: (i) uzyskanie informacji dotyczących struktury, właściwości fizycznych oraz mechanizmów tworzenia i rozpadu jonów organicznych[53,59], w tym jonów alkilofenyloamoniowych[33], karbeniowych i immoniowych[53]; (ii) obserwację reakcji przeniesienia wodoru[53,61] i
przegrupowania szkieletowego[62,63]; (iii) wyznaczenie sekwencji
aminokwasów w peptydach i białkach[34,35] oraz w glikopeptydach[64];
(iv) określenie budowy i stabilności kompleksów metali przejściowych[30-32],
strukturalnych i stereoizomerów steroidów[29]; (v) wyznaczenie energii
wiązań[60,63,65] i zasadowości w fazie gazowej cząsteczek organicznych[63];
procesie kationów metali alkalicznych i kationu amoniowego[36]; (vii)
analizę składu mieszanin związków[44,46,48,59].
Technika CID istnieje w trzech odmianach[68]: (i) tandemowa
spektrometria mas, (ii) CID przeprowadzana w źródle jonów oraz (iii) CID przeprowadzana w pułapce jonowej (ang. ion trap, IT) lub komorze spektrometru typu spektrometru jonowego rezonansu cyklotronowego z transformacją Fouriera (ang. Fourier transform ion cyclotron resonance, FTICR). Ta ostatnia odmiana CID posiada duże możliwości analityczne w badaniu równowag reakcji w fazie gazowej, wyznaczaniu ich parametrów kinetycznych i termodynamicznych[63], lecz w dalszym omówieniu
szczegółów dysocjacji zderzeniowej zostanie pominięta ze względu na odmienność zachodzących w niej procesów i stosowanych rozwiązań konstrukcyjnych.
Tandemowa spektrometria mas (ang. tandem mass spectrometry, MS/MS lub MSn), łączy wszystkie techniki, w których wybrany w
analizatorze mas jon (M1) ulega powtórnej analizie mas (M2)[19,31]. Gdy
spektrometr mas jest wyposażony w kilka złączonych kolejno analizatorów, istnieje możliwość odseparowania interesującego jonu za pomocą pierwszego analizatora, przeprowadzenia kontrolowanych zderzeń w komorze kolizyjnej i analizy jonów, powstałych na skutek CID, za pomocą kolejnego analizatora mas. Kontrola energii wewnętrznej jonu, trafiającego do komory zderzeń, odbywa się w takim układzie poprzez kontrolę ciśnienia gazu kolizyjnego oraz energii kinetycznej jonu[37]: podwyższenie ciśnienia
gazu powoduje zwiększenie energii wewnętrznej jonu poprzez zwiększenie prawdopodobieństwa zderzeń pomiędzy jonem i cząsteczkami gazu[37]. Z
kolei energia kinetyczna interesującego jonu, opuszczającego pierwszy analizator mas może ulegać dodatkowemu zwiększeniu (gdy takim analizatorem jest analizator kwadrupolowy, dla którego charakterystyczne jest małe przyśpieszanie jonów podczas analizy)[19,63] lub zmniejszeniu (gdy
pierwszym analizatorem jest analizator elektryczno-magnetyczny, silnie przyśpieszający jony podczas analizy)[33,35,63]. Ciśnienie gazu w komorze
zderzeń utrzymuje się w granicach 10-6 - 10-9 Pa [53], co powoduje, że CID
jest przeprowadzana w warunkach pojedyńczego zderzenia interesującego jonu z cząsteczką gazu[19,33,35]. To pozwala na dokładną ocenę ilości energii, przekształconej w energię wewnętrzną jonu[63], co umożliwia dokładne i
wiarygodne oznaczenie np. energii wiązań[63,65].
Drugą odmianą CID jest dysocjacja zderzeniowa w źródle jonów, występująca w kilku odmianach, różniących się rozwiązaniem konstrukcyjnym układu ogniskującego źródła jonów (rys. 3) (ang. skimmer
CID[19,66], skimmer cone CID[33], sCID[66], in-source fragmentation[11,35,38],
in-source CID[33], cone-voltage fragmentation[12,33,35], cone-voltage CID[33],
nozzle-skimmer dissociation[12], nozzle-skimmer fragmentation[33,35],
nozzle/skimmer CID[11,19], high orifice potential fragmentation[33,35], up-front
CID[29]). Ciśnienie gazu kolizyjnego w obszarze stożkowego separatora
jonów [(1 – 7)×10-2 Pa] jest wystarczająco niskie, żeby zapewnić jonom
odpowiedni tor ruchu przed kolizją, lecz wystarczająco duże, żeby prawdopodobieństwo wielokrotnych kolizji było wysokie. Chociaż względnie wysokie ciśnienie gazu w tym obszarze może w większym stopniu spowodować utratę przez jon energii kinetycznej poprzez kolizje [Estr, w równ. (1)], niż jego rozpad, to jednak odpowiednio zmieniając
różnicę potencjałów pomiędzy częściami źródła jonów (w zależności od konstrukcji źródła jonów, zwiększa się wartość napięcia przykładanego do otworów albo stożkowych separatorów jonów, których w niektórych układach może występować kilka)[67], można nie tylko osiągać pełną
desolwatację jonu[11,19,37], lecz także zmieniać jego energię kinetyczną
[składnik q∆V w równ. (1)][34,35]. Najczęściej zmienia się różnicę potencjałów pomiędzy stożkowym separatorem jonów a otworem (potencjał
stożkowy, ang. cone voltage[30,20], cv). Przy wartości potencjału stożkowego
równej zeru, jon posiada tylko energię termiczną, której bardzo mała ilość jest przetwarzana na energię wewnętrzną jonu. Z tego powodu nie zachodzą desolwatacja i fragmentacja jonu, a transmisja jonu w kierunku analizatora jest bardzo słaba[34,37]. Poprzez zmianę natężenia pola elektrycznego pomiędzy otworem i stożkowym separatorem jonów można zatem zmieniać energię wewnętrzną interesującego jonu i przeprowadzać jego kontrolowaną fragmentację według mechanizmu CID[11,19,29-31,33,37].
kapilara
elektroda przeciwnie naładowana
otwór (ang. orifice, nozzle)
stożkowy separator jonów (ang. skimmer, cone) elektrostatyczny układ ogniskujący
10 Pa (1 - 7) N2
soczewki elektrostatyczne
10-2 Pa
Rys. 3. Schemat źródła jonów ES. Ciśnienia podane według Schneider i Chen[37].
Zgodnie z równ. (1), gaz kolizyjny o większej masie cząsteczkowej (np. N2[30,31,33,68]) i większe różnice potencjału stożkowego powodują
ekstensyfikację i intensyfikację reakcji fragmentacji[11,37,66]. Ze względu na
złożoność procesów aerodynamicznych[37], zachodzących w obszarze
stożkowego separatora jonów, obecnie trudna jest ilościowa ocena części energii, przekształcanej podczas kolizji w energię wewnętrzną jonu, jednak
![Rysunek 3. Pochodne makrocyklicznych diamin [12]-N 2 O 2 , [15]-N 2 O 3 i [18]-N 2 O 4](https://thumb-eu.123doks.com/thumbv2/9liborg/3118877.8947/103.892.186.659.723.920/rysunek-pochodne-makrocyklicznych-diamin-n-o-n-o.webp)