1
Mgr inż. biotechnologii Marianna TyczewskaPROFIL EKSPRESJI WYBRANYCH GENÓW ZWIĄZANYCH ZE WZROSTEM,
RÓŻNICOWANIEM I CZYNNOŚCIĄ KOMÓREK KORY NADNERCZA SZCZURA
W PRZEBIEGU INDUKOWANEJ ENUKLEACJĄ REGENERACJI GRUCZOŁU.
Rozprawa doktorska
Promotor: prof. dr hab. Ludwik K. Malendowicz
Katedra I Zakład Histologii I Embriologii, Uniwersytet Medyczny im. Karola Marcinkowskiego w Poznaniu
2
Za okazaną pomoc, wsparcie i życzliwość w trakcie przygotowywania niniejszej rozprawy składam Panu Profesorowi Ludwikowi K. Malendowiczowi serdeczne podziękowania.
Dziękuję bardzo Panu Profesorowi Maciejowi Zablowi, kierownikowi Katedra i Zakładu Histologii i Embriologii UM w Poznaniu za stworzenie wyjątkowej, sprzyjającej pracy
naukowej atmosfery w Zakładzie.
Dziękuję serdecznie Panu Profesorowi Andrzejowi Łukaszykowi za okazaną pomoc w trakcie prowadzenia badań.
3
Część wyników badań ujętych w niniejszej rozprawie opublikowano jako:
Tyczewska, M., Rucinski, M., Trejter, M., Ziolkowska, A., Szyszka, M., Malendowicz, L.K.: Angiogenesis in the course of enucleation-induced adrenal
regeneration--expression of selected genes and proteins involved in development of capillaries. Peptides 38: 404-413 (2012) IF: 2.434, punktacja MNiSW: 25.000
Tyczewska, M., Rucinski, M., Ziolkowska, A., Trejter, M., Szyszka, M., Malendowicz, L.K.: Expression of selected genes involved in steroidogenesis in the course of
enucleation-induced rat adrenal regeneration. Int J Mol Med 33: 613-623 (2013) IF: 1.957, punktacja MNiSW: 20.000
4
SPIS TREŚCI
Skróty stosowane w pracy Str. 6
1. Wstęp Str. 7
1.1. Indukowana obustronną enukleacją regeneracja nadnerczy jako
jeden z modeli eksperymentalnej endokrynologii Str. 7 1.2. Proliferacja komórek regenerującej kory nadnerczy Str. 8 1.3. Angiogeneza w świetle regeneracji kory nadnercza szczura Str. 9 1.4. Steroidogeneza w toku indukowanej enukleację regeneracji kory
nadnercza szczura Str. 10
2. Założenia i cel pracy Str. 12
3. Materiały i metody Str. 13
3.1. Materiały Str. 13
3.1.1. Zwierzęta i opis doświadczeń Str. 13
3.1.2. Badania histologiczne Str. 13
3.1.3. Odczynniki (z nr katalogowym, jeśli dostępny) Str. 13
3.1.4. Strony internetowe, oprogramowanie Str. 15
3.2. Metody Str. 15
3.2.1. Izolacja całkowitego RNA Str. 15
3.2.2. Metoda mikromacierzy RNA Str. 16
3.2.2.a. Przygotowanie kontrolnego i całkowitego RNA Str. 16 3.2.2.b. Synteza pojedynczo-niciowego cDNA Str. 17
3.2.2.c. Transkrypcja in vitro Str. 17
3.2.2.d. Oczyszczanie cRNA oraz ocena ilościowo-jakościowa Str. 18
3.2.2.e. Drugi cykl cDNA Str. 18
3.2.2.f. Hydroliza, oczyszczanie oraz ocena
ilościowo-jakościowa materiału Str. 19
3.2.2.g. Fragmentacja cDNA Str. 20
3.2.2.h. Znakowanie cDNA Str. 20
3.2.2.i. Hybrydyzacja Str. 20
3.2.2.j. Płukanie macierzy i odczyt wyników Str. 21 3.2.2.k. Analiza danych uzyskanych metodą mikromacierzy
RNA Str. 23
3.2.3. Walidacja wyników otrzymanych metodą mikromacierzy RNA
za pomocą metody QPCR Str. 25
3.2.3.a. Odwrotna transkrypcja Str. 25
3.2.3.b. QPCR Str. 25
3.2.3.c. Przygotowanie krzywej standardowej Str. 27
3.2.4. Badanie immunohistochemiczne Str. 28
3.2.4.a. Dwustopniowa metoda immunoperoksydazowa z
kompleksem EnVision-HRP Str. 28
5
3.2.5. Analiza statystyczna wyników Str. 29
4. Wyniki Str. 30
4.1. Profil globalny ekspresji genów w toku indukowanej enukleacją
regeneracji nadnerczy szczura Str. 30
4.2. Angiogeneza w indukowanej enukleacją regeneracji nadnercza
szczura Str. 33
4.2.1. Analiza ekspresji genów związanych z angiogenezą Str. 33 4.2.2. Walidacja metodą QPCR ekspresji wybranych genów Str. 38 4.2.3. Lokalizacja immunohistochemiczna produktów wybranych
genów związanych z angiogenezą Str. 40
4.3. Proliferacja w indukowanej enukleacją regeneracji nadnercza
szczura Str. 42
4.3.1. Analiza ekspresji genów związanych z proliferacją Str. 42
4.3.2. Walidacja metodą QPCR ekspresji genów Str. 46
4.4. Steroidogeneza w indukowanej enukleacją regeneracji nadnercza
szczura Str. 49
4.4.1. Analiza ekspresji genów związanych ze steroidogenezą Str. 49 4.4.2. Walidacja metodą QPCR ekspresji wybranych genów Str. 53 4.4.3. Lokalizacja immunohistochemiczna produktów wybranych
genów związanych ze steroidogenezą Str. 56
5. Dyskusja Str. 58
6. Wnioski Str. 74
7. Streszczenie Str. 75
6
Skróty stosowane w pracyACTH hormon adrenokortykotropowy, adrenokortykotropina Adam A disintegrin and metalloprotease
Asp nadnerczowa proteaza serynowa (adrenal serine protease) cAMP cykliczny adenozynomonofosforan
Crh kortykoliberyna (CRF)
DAB chlorowodorek diaminobenzydyny DNA kwas dezoksyrybonukleinowy HPA oś podwzgórze-przysadka-nadnercze Hprt fosforybozylo-transferaza hipoksantyny. ICH immunohistochemia
mRNA matrycowy RNA
PBS bufor fosforanowy w soli fizjologicznej PCR reakcja łańcuchowa polimerazy DNA POMC proopiomelanokortyna
QPCR reakcja PCR w czasie rzeczywistym Tgf α transformujący czynnik wzrostu α Tgfb transformujący czynnik wzrostu β Tnf-α czynnik martwicy nowotworów α ZF strefa pasmowata kory nadnercza ZG strefa kłębkowata kory nadnercza Cyp11a1 desmolaza cholesterolowa (P450scc) Hsd3b dehydrogenaza 3beta-hydroksysteroidowa Cyp11b1 11beta-hydroksylaza
Cyp11b2 syntaza aldosteronu
Vegf śródbłonkowy czynnik wzrostu
Fgf czynnik wzrostu fibroblastów Pdgf płytkowy czynnik wzrostu TH hydroksylaza tyrozynowa
aFgf kwasowy czynnik wzrostu fibroblastów bFgf zasadowy czynnik wzrostu fibroblastów Il-1 interleukina 1
Il-6 interleukina 6 Il-8 interleukina 8
Vwf czynnik von Willebranda Ang-1 angiopoetyna 1
Ang-2 angiopoetyna 2
Thy1 (CD90) glikoproteina powierzchniowa Tie2 receptor angiopoetyny
SR-B1 receptor Scarb1
HDL lipoproteiny wysokiej gęstości LDL lipoproteiny niskiej gęstości Lipe lipaza
acetylo-CoA acetylo-koenzymu A
7
1. Wst
ęp
1.1. Indukowana obustronną enukleacją regeneracja nadnerczy jako jeden z modeli
eksperymentalnej endokrynologii.
Jednym z najczęściej stosowanych modeli badań nad kontrolą wzrostu nadnerczy w warunkach in vivo jest indukowana obustronną enukleacją regeneracja nadnerczy szczura. Zabieg ten polega na usunięci zawartości nadnerczy - zarówno kory jak i rdzenia (Ingle i Higgins, 1938). W narządzie pozostają komórki przylegające do torebki łącznotkankowej, z których następuje regeneracja gruczołu. W literaturze proces regeneracji nadnercza opisywany jest jako przypominający wczesne etapy ontogenezy (Holzwarth i wsp., 1980; Taki i Nickerson, 1985; Zieleniewski i Jankiewicz, 1986; Nowakowska-Jankiewicz i wsp., 1998). Proces ten związany jest z odbudową kory nadnercza i obejmuje zarówno hiperplazję komórek miąższowych, jak i ich hipertrofię i jest związany z różnicowaniem (Dallamn, 1984-85; Taki i Nickerson, 1985). Enukleacja nadnercza prowadzi do bardzo szybkiej odbudowy kory. Na podstawie zmian masy regenerujących nadnerczy i ilości w nich DNA stwierdzono, że w przeciągu około 3-4 tygodni struktura kory zostaje odbudowana i gruczoł w pełnić odzyskuje swoje funkcje (Holzwarth i wsp., 1980; Zieleniewski i Nowakowska-Jankiewicz, 1986; Engeland i wsp., 1996; Miyamoto i wsp., 2000; Ennen i wsp., 2005; Kim i Hammer, 2007; Tyczewska i wsp., 2012, 2014).
Impulsem do regeneracji kory nadnerczy jest spadek poziomu kortykosteronu we krwi, a podawanie kortykosteronu hamuje regenerację. Nagły spadek poziomu kortykosteronu we krwi wywołuje kompensacyjny wzrost poziomu krążącego ACTH oraz N-końcowych peptydów proopiomelanokortyny (N-POMC) (Estivariz i wsp., 1982, 1988; Perone i wsp., 1997). Peptydy powstające z POMC odgrywają decydującą rolę w procesie regeneracji, gdyż hipofizektomia powoduje zahamowanie regeneracji (Buckingham i wsp., 1975; Alfano i wsp., 1985; Estivariz i wsp., 1988). Pomiędzy 10. a 15. dniem regeneracji poziom ACTH we krwi jest dwa razy wyższy w porównaniu z kontrolą, natomiast nie stwierdza się w tym okresie znaczących różnic w poziomie kortykosteronu. Dopiero około 20. dnia regeneracji, poziom kortykosteronu we krwi wzrasta i osiąga wartości porównywalne z kontrolą. Wtedy także w surowicy krwi obniża się poziom ACTH, co wskazuje na odbudowę w tym okresie funkcjonalnego sprzężenia zwrotnego pomiędzy podwzgórzem, przysadką mózgową a nadnerczami (Ennen i wsp., 2005).
8
Źródłem regenerujących komórek kory nadnerczy są komórki macierzyste kory gruczołu, zlokalizowane głównie podtorebkowo, którym towarzyszą pozostałe po zabiegu komórki strefy kłębkowatej kory gruczołu (Taki i Nickerson, 1985; Mitani i wsp., 1994, 1995; Ennen i wsp., 2005). Przyjmuje się, że komórki macierzyste podobne są do komórek strefy pośredniej, które w normalnym nadnerczu są komórkami ściśle upakowanymi, o niewielkiej liczbie kropli lipidowych, umiejscowionymi pomiędzy strefą ZG i ZF. Wykazują one ekspresję mRNA desmolazy cholesterolowej (Cyp11a1) i dehydrogenazy 3β-hydroksysteroidowej (Hsd3b) - enzymów zaangażowanych we wczesne etapy steroidogenezy (Engeland i Leway-Young, 1999). Wykazano, iż komórki strefy pośredniej posiadają zdolność różnicowania w komórki wszystkich stref kory nadnerczy, a więc stanowią one grupę oligopotentnych komórek macierzystych (Mitani i wsp., 1994, 1995, 2003). Wywodzące się z nich klony komórek migrują centropetalnie - od okolicy podtorebkowej do środka gruczołu, czyli do najgłębszych części strefy siatkowatej.Regeneracja kory nadnercza związana jest z wysokim stopniem proliferacji zarówno komórek miąższowych jak i komórek naczyń krwionośnych. Od pierwszych dni regeneracji trwa systematyczny wzrost liczby komórek parenchymalnych (Taki i Nickerson, 1985). Jak już wcześniej wspomniano, proces odbudowy kory gruczołu hamowany jest podaniem kortykosteroidów lub pozostawieniem jednego nadnercza nienaruszonego (Skelton, 1958; Holzwarth i wsp., 1980; Engeland i wsp., 1999). Całkowite usunięcie nietkniętego nadnercza nawet kilka tygodni po zabiegu enukleacji, pobudza regenerację nadnercza enukleowanego (Engeland i wsp., 2005).
Tempo oraz zakres zmian w trakcie regeneracji nadnerczy związane są głównie z odbudową strefy pasmowatej (zona fasciculata, ZF) kory, której struktura i funkcja odbudowuje się już przed 20. dniem regeneracji, podczas gdy odbudowa strefy kłębkowatej (zona glomerulosa, ZG) przed 30. dniem regeneracji pozostaje nieukończona (Engeland i Levay-Young, 1999).
1.2. Proliferacja komórek regenerującej kory nadnerczy.
Bezpośrednio po enukleacji w nadnerczu szczura dochodzi do powstania skrzepu, który równocześnie indukuje proces zapalny w narządzie. Jednocześnie na tym etapie następuje inicjacja proliferacji komórek miąższowych kory gruczołu, w trakcie której komórki przygotowują się do podziałów komórkowych. U szczura już pod koniec 3. dnia regeneracji
9
obserwuje się wyraźną, intensywną proliferację komórek miąższowych kory nadnerczy, trwającą do siódmego dnia po zabiegu (Taki i Nickerson, 1985). Między 8. a 10. dniem regeneracji pojawiają się już pierwsze elementy histologicznej organizacji narządu, przede wszystkim strefa pasmowata, gdyż na tym etapie regeneracji nie można rozpoznać ani strefy kłębkowatej, ani też siatkowatej. Co więcej, komórki miąższowe kory nadnerczy na tym etapie regeneracji nie wykazują ekspresji mRNA syntazy aldosteronu (Cyp11b2), markera strefy kłębkowatej (Engeland i Leway-Young, 1999).Uważa się, że także rdzeń nadnercza ulega regeneracji, jednakże proces ten jest znacznie dłuższy i nie doprowadza do powstania rdzenia gruczołu jako jednolitej centralnie położonej struktury. Komórki chromafinowe występują jako pojedyncze komórki rozrzucone pomiędzy komórkami steroidogennymi (Kmieć, 1968). Już we wczesnym etapie regeneracji (3-7. dzień regeneracji) wykazano obecność pojedynczych komórek wykazujących odczyn na hydroksylazę tyrozynową (komórki TH-pozytywne), a występują one głównie w obszarze podtorebkowym. Komórki te następnie prawdopodobnie migrują do centralnej części regenerującego nadnercza (14. dzień regeneracji) (Ulrich-Lay i Engeland, 2000).
1.3. Angiogeneza w świetle regeneracji kory nadnercza szczura.
Proces powstawania nowych naczyń krwionośnych odgrywa istotną rolę zarówno w życiu płodowym, jak i dorosłym. W dorosłym organizmie angiogeneza zachodzi rzadko, lecz odgrywa istotną rolę w reprodukcji, patogenezie, procesach naprawczych tkanek i procesach zapalnych (Folkman, 1995; Lee i wsp., 1998; Risau, 1997).
Jednym z modeli badań nad procesem angiogenezy jest regeneracja narządów. Pod tym względem, rozwój unaczynienia musi być skoordynowany ze wzrostem narządu (Ishimoto i wsp., 2006; Thomas i wsp., 2003). Na przykład, w trakcie regeneracji wątroby, powstawanie nowych naczyń włosowatych z już istniejących poprzez rozgałęzianie jest warunkiem koniecznym proliferacji hepatocytów (Taniguchi i wsp., 2001). Proces jest kontrolowany przez czynnik wzrostu śródbłonków (Vegf), którego mRNA ulega ekspresji w hepatocytach oraz pozostałych komórkach wątroby (Ishikawa i wsp., 1999; Mochida i wsp., 1996).
Jak wcześniej wspomniano, warunkiem gwałtownej proliferacji komórek kory nadnercza prowadzącej do odbudowy struktury gruczołu w toku indukowanej enukleacją regeneracji kory nadnerczy jest odbudowa sieci naczyń krwionośnych w obrębie gruczołu. Obok bardzo
10
dobrze znanych czynników wzrostu, które kontrolują angiogenezę (indukują lub hamują ją) - przykładowo Vegf, kwasowy/zasadowy czynnik wzrostu fibroblastów (aFgf, bFgf), angiopoetyny (Ang-1, Ang-2), transformujący czynnik wzrostu beta (Tgfb), czynnik von Willebranda (Vwf) - również wiele cytokin (np., interleukina 8, Il-1, Il-6) pośrednio wpływa na angiogenezę poprzez indukcję ekspresji różnych receptorów błonowych (Ferrera, 1999; Frater-Schroder i wsp., 1987; Pepper i wsp., 1996). Jedną z nich jest Thy1 (CD90), glikoproteina powierzchniowa o masie 25-37 kDa, pierwotnie opisana jako marker różnicowania tymocytów u myszy, która, jak się okazało, związana jest z proliferacją oraz różnicowaniem różnych typów komórek (Schubert i wsp., 2011). Ponadto, ostatnio zidentyfikowano czynniki zaangażowane w regulację angiogenezy, którymi są angiopoetyny (Ang-1 i Ang-2). Te niedawno odkryte ligandy receptorów Tie2 specyficznych dla komórek śródbłonka, ulegają też ekspresji w komórkach nadnerczy i są regulowane przez ACTH (Feraud i wsp., 2003). System Ang-1, Ang-2 i receptory Tie2, w porównaniu do Vegf, powiązany jest z dojrzewaniem, stabilizacją i funkcjonowaniem systemu naczyń krwionośnych.W przypadku nadnerczy, rozwój unaczynienia oraz masy komórek miąższowych jest także kontrolowany przez ACTH (Thomas i wsp., 2003). Kortykotropina wpływa na unaczynienie i reguluje ekspresję różnych czynników angiogenezy, m.in. Vegf.
1.4. Steroidogeneza w toku indukowanej enukleację regeneracji kory nadnercza
szczura.
Podstawową funkcją komórek kory nadnercza jest produkcja hormonów steroidowych. Podstawowym substratem w syntezie steroidów jest wolny cholesterol, który powstaje głównie z estrów cholesterolu na drodze hydrolizy. Pozyskiwanie estrów cholesterolu może odbywać się przy udziale receptora Scarb1 (SR-B1), który wiąże lipoproteiny wysokiej gęstości (HDL), a następnie w cytoplazmie komórek, przy udziale specyficznej lipazy (Lipe) są one przekształcane w wolny cholesterol. Z drugiej strony, cholesterol z lipoprotein niskiej gęstości (LDL) pobierany jest na drodze endocytozy z udziałem receptorów, a dalej hydrolizowany przez kwaśną esterazę w lizosomach (Balasubramaniam i wsp., 1977; Nishikawa i wsp., 1981; Gwynne i Mahaffee, 1989; Li i wsp., 2002; Azhar i Reaven, 2002; Connelly i Williams, 2004; Miller i Bose, 2011). Estry cholesterolu magazynowane są w kroplach lipidowych. Ponadto, wolny cholesterol dostarczany jest także na drodze syntezy de novo z acetylo-koenzymu A (acetylo-CoA) na siateczce śródplazmatycznej przy udziale
11
acetylotransferazy (Soat1), jednak w ten sposób powstaje zaledwie około 20% wolnego cholesterolu (Kraemer, 2007). U gryzoni głównym źródłem wolnego cholesterolu są krążące cząsteczki HDL. Zarówno hydroliza cholesterolu, jak i synteza sterydów podlegają kontroli ACTH (Trzeciak i wsp., 1974; Malendowicz, 1986; Schimmer i wsp., 2006).Jak wcześniej wykazano, pozostawione pod torebką po enukleacji nadnerczy komórki nie wykazują ekspresji ani syntazy aldosteronu, ani też 11 beta-hydroksylazy (Engeland i Levay-Young, 1999). Oba enzymy są markerami odpowiednio komórek strefy kłębkowatej (ZG) i pasmowatej (ZF). Z tego też względu, do niedawna uważano, że komórki regenerującej kory nadnercza powstają z odróżnicowanych komórek strefy ZG pozostawionych pod torebką podczas zabiegu enukleacji, których fenotyp podobny jest do fenotypu komórek strefy pośredniej, stanowiących komórki macierzyste gruczołu (Mitani i wsp., 1994, 1995, 2003; Engeland i Levay-Young, 1999).
Jak już wcześniej wspomniano, enukleacja nadnerczy prowadzi do gwałtownego spadku poziomu kortykosteronu we krwi, co indukuje kompensacyjną hipersekrecję ACTH. Regeneracja jest hamowana, gdy jedno z nadnerczy pozostaje nietknięte, a stężenie kortykosteronu w surowicy krwi utrzymuje się na normalnym poziomie i w tych warunkach uwalnianie ACTH podlega sprzężeniu zwrotnemu (Engeland i Levay-Young, 1999; Holzwarth i wsp., 1980; Buckingham i Hodges, 1974). Również podawanie hormonów steroidowych zapobiega regeneracji kory nadnerczy. Wcześniejsze badania wykazały, że podczas regeneracji nadnerczy ekspresja zarówno syntazy aldosteronu, jak i 11beta-hydroksylazy pozostaje obniżona podczas pierwszego tygodnia regeneracji (hybrydyzacja in situ). Należy zaznaczyć, że poziom ekspresji Cyp11b2 pozostaje niski w porównaniu do kontroli nawet przed 30. dniem regeneracji nadnerczy, podczas gdy ekspresja Cyp11b1 i Hsd3b wraca do poziomu porównywalnego do kontroli około 20. dnia po enukleacji (Engeland i Levay-Young, 1999).
12
2. Zało
żenia i cel pracy
Z powyższego krótkiego przeglądu piśmiennictwa dotyczącego kluczowych procesów zachodzących w toku indukowanej enukleacją regeneracji nadnercza szczura wynika, iż szereg zjawisk związanych z odbudową struktury i funkcji nadnercza nie jest wyjaśnionych. Dlatego celem przeprowadzonych badań było:
♦ określenie zmian profilu globalnej ekspresji genów w toku indukowanej enukleacją regeneracji nadnercza szczura oraz wyodrębnienie z nich, w oparciu o programy komputerowe – analiza klasteryzacji oraz baza GO, różnych grup funkcjonalnych,
♦ szczegółowa analiza w regenerującym nadnerczu ekspresji genów związanych z procesem angiogenezy (wyodrębnienie genów o największych zmianach w ekspresji w porównaniu z grupą kontrolną), walidacja ich ekspresji przy pomocy QPCR oraz lokalizacja immunohistochemiczna produktów białkowych wybranych genów w regenerującym nadnerczu,
♦ szczegółowa analiza w regenerującym nadnerczu ekspresji genów związanych z podstawowymi funkcjami komórek – wzrostem i proliferacją (wyodrębnienie genów o największych zmianach w ekspresji w porównaniu z grupą kontrolną), walidacja ich ekspresji przy pomocy QPCR,
♦ szczegółowa analiza w regenerującym nadnerczu ekspresji genów związanych z procesem steroidogenezy (wyodrębnienie genów o największych zmianach w ekspresji w porównaniu z grupą kontrolną), walidacja ich ekspresji przy pomocy QPCR oraz lokalizacja immunohistochemiczna produktów białkowych wybranych genów w regenerującym nadnerczu.
13
3. Materiały i metody
3.1. Materiały
3.1.1. Zwierzęta i opis doświadczeń
Protokoły przedstawionych doświadczeń uzyskały zgodę Lokalnej Komisji Etycznej do Spraw Doświadczeń na Zwierzętach nr 19/2011.
Doświadczenia przeprowadzono na samicach szczura szczepu Wistar (ok. 100 g), które pochodziły z hodowli Katedry i Zakładu Toksykologii, Uniwersytetu Medycznego w Poznaniu. Przed rozpoczęciem doświadczeń i w trakcie ich trwania szczury przetrzymywano w zwierzętarni Katedry i Zakładu Histologii i Embriologii Uniwersytetu Medycznego w Poznaniu, w warunkach standardowych (temperatura 22±2 oC, oświetlenie 14L/10D), przy stałym dostępie do wody i paszy laboratoryjnej.
W trakcie przeprowadzonego zabiegu enukleacji, szczury poddawano uśpieniu podając ketaminę i.m. w dawce 1 mg/100 g masy ciała szczura oraz ksylaminę i.m. w dawce 0,2 mg/100 g masy ciała szczura.
Na wysokości ostatniego żebra, z nacięcia grzbietowego przeprowadzono obustronną enukleację nadnerczy, która polega na przecięciu torebki łącznotkankowej, usunięciu kory i rdzenia nadnerczy, z pozostawieniem torebki łącznotkankowej i przylegających do niej komórek. Doświadczenie przeprowadzono między godzinami 10:30 a 11:30. Przez pierwsze 3 doby szczurom podawano do picia roztwór soli fizjologicznej. Pierwszą sekcję przeprowadzono po upływie 24 godzin od enukleacji, kolejne sekcje po upływie kolejnych dób (2., 3., 5., 8. oraz 15.). Kontrolę stanowiły nadnercza pobrane od 6-tygodniowych szczurów, które poddano operacji pozornej, tzn. rozcięcie skóry, rozwarstwienie tkanki mięśniowej i zszycie skóry. Materiał od tych zwierząt pobierano po 24 godz. od zabiegu.
3.1.2. Badania histologiczne
Materiał do badań stanowiły, pobierane bezpośrednio po dekapitacji, nadnercza, które oczyszczano z przylegającej tkanki tłuszczowej, przepłukiwano je w roztworze PBS) i umieszczano w probówkach zawierających RNA-later lub płyn Bouina (liczba nadnerczy w każdej próbce n=6). Próbki przeznaczone do badań molekularnych (mikromacierze RNA oraz QPCR) przechowywano w zamrażarce w temperaturze -20 oC.
3.1.3. Odczynniki (z nr katalogowym, jeśli dostępny)
14
Zestaw do znakowania (Ambion WT Expression Kit, WT Terminal Labeling and ControlsKit)
Bromek etydyny (Sigma – Aldrich, A-3678)
DNA ladder (wzorzec wielkości DNA, Fermentas, SM0613) DNA-za Rnase – Free Dnase Set (Qiagen, 79254)
Isopropanol (Sigma – Aldrich, I- 9516)
Lightcycler FastStart DNA Master SYBR Green I (Roche) Mikromacierze RNA (Affimetrix Array Strip, Affimetrix) Zestaw do hybrydyzacji (Hybridization Control Kit, Affymetrix)
Zestaw do hybrydyzacji (Hybridization, Wash, and Stain Kit, Affymetrix)
Zestaw kontrolny do mikromacierzy RNA (Poly-A RNA Control Kit, Affymetrix) Probówki 1.5 ml, 2 ml (Eppendorf)
RNA later (Qiagen, 1018087)
RNA zap (Sigma – Aldrich, R-2020) Rneasy RNA extraction kit (Qiagen)
TRI Reagent (TRI-zol, Sigma – Aldrich, T-9424) Trizma base (Sigma – Aldrich, T6066)
PBS
Prime RNase Inhibitor, 7 500 u (Eppendorf, 0032 005 357) Przeciwciało anty Star (ab58013, abcam®)
Przeciwciało anty Hsd11b (ab39364, abcam®) Przeciwciało anty Cyp11a1 (bs-3608R, Bioss®) Przeciwciało anty Cyp11b1 (bs-3898R, Bioss®) Przeciwciało anty Vegfa (Thermo SCIENTIFIC) Przeciwciało anty Thy-1 (MRC OX-7, ab225, abcam®) Przeciwciało anty CD34 (abcam®)
Przeciwciało przeciwko króliczemu IgG związane z peroksydazą chrzanową (Sigma, St. Louis, MO, USA)
Surowica kozia normalna (Sigma, X-0907)
Zestaw do izolacji RNA NucleoSpin RNA II (Macherey-Nagel, 740 955.250) Przeciwciało anty CD34 (Abcam, ab64480)
Przeciwciało anty Kdr (Vegfr2) (Abcam, ab2349) bufor cytrynianowy pH 6 (Dako, S1699)
15
EnVision + System-HRP (DAB) (Dako, K4011)Pozostałe odczynniki zakupiono w POCh Gliwice, cz.d.a.
3.1.4. Strony internetowe, oprogramowanie.
Blast (porównywanie homologii sekwencji) - http://www.ncbi.nlm.nih.gov/BLAST
Primer 3 (projektowanie starterów do reakcji PCR) - http://www.genome.wi.mit.edu/cgi bin/primer/primer3.cgi
ClustalW (analiza sekwencji nukleotydowych oraz białkowych)
Statistica (analiza statystyczna uzyskanych wyników)
Genorm (wyszukiwanie najbardziej stabilnego zestawu genów referencyjnych do normalizacji wyników Q-PCR)
http://www.affymetrix.com/estore/index.jsp
bioconductor (pakiet analizy wyników mikromacierzowych języka programistycznego R)
geneanswers (pakiet do geupowania genów o zmiennej ekspresji według opisu z bazy GeneOntology)
3.2. Metody.
3.2.1. Izolacja całkowitego RNA.
Izolację całkowitego RNA ze wszystkich pobranych nadnerczy przeprowadzono metodą standardową za pomocą odczynnika TRI Reagent. Ze względu na wysoką wrażliwość RNA na degradację, proces ten przeprowadzono w komorze laminarnej, której powierzchnię potraktowano etanolem oraz dodatkowym odczynnikiem RNA-zap. Tkanki rozmrażano, usuwano RNA-later poprzez kilkakrotne płukanie tkanek w wodzie wolnej od nukleaz, a następnie poddawano homogenizacji w 1 ml odczynnika TRI Reagent. Całość przenoszono do czystych probówek, dodawano po 200 µl chloroformu (200 µ l chloroformu na każde 500 µ l mieszaniny), energicznie wytrząsano przez 15 sekund, po czym inkubowano w temperaturze pokojowej przez 10 – 15 minut, a następnie wirowano w 12000x g przez 15 minut w temperaturze 4 oC. W wyniku wirowania każda próbka rozdzielona została na trzy fazy (dolną, środkową i górną). Tylko górna faza zawierała RNA, którą zbierano do czystych probówek. W celu wytrącenia RNA dodawano 0,5 ml izopropanolu. Całość inkubowano przez godzinę w temperaturze –20 oC, następnie wirowano przy 12000x g przez 20 minut w
16
temperaturze 4 oC, po czym usuwano supernatant i dodawano 1 ml 70% etanolu. Przez 20 sekund mieszaninę intensywnie wytrząsano. Mieszaninę wirowano przy 12000x g przez 15 minut w temperaturze 4 oC. Kilkukrotnie powtarzano przemywanie etanolem. Uzyskany osad RNA osuszano, po czym rozpuszczano w 30 µl wody wolnej od nukleaz. Pozostałości DNA usuwano za pomocą trawienia DNA-zą w temperaturze pokojowej przez 30 minut.. Otrzymane w taki sposób RNA oczyszczano z mieszaniny reakcyjnej poprzez wirowanie na kolumienkach, a związane z membraną kolumienek RNA wypłukiwano poprzez dodanie 20 µ l wody wolnej od nukleaz. Stężenie uzyskanego RNA mierzono spektrofotometrycznie (NanoDrop Technologies), a jego jakość sprawdzano poprzez rozdział na żelu agarozowym 1.5% z dodatkiem bromku etydyny.Po ukończeniu izolacji całkowitego RNA, część materiału pobrano w celu analizy ekspresji genów metodą mikromacierzy ekspresyjnych, natomiast resztę zamrożono w temperaturze -80 oC w celu późniejszej walidacji wyników metodą QPCR.
3.2.2. Metoda mikromacierzy RNA.
Całą procedurę badania ekspresji genów za pomocą mikromacierzy RNA przeprowadzono przy użyciu systemu firmy Affymetrix – GeneAtlas™ Personal Microarray System. Przebadano blisko 30 tysięcy genów szczura, wykorzystując mikromacierze RNA typu GeneChip® Rat Gene ST Arrays (Affymetrix® Gene 1.1 ST Array Strip for Rat).
3.2.2.a. Przygotowanie kontrolnego i całkowitego RNA.
W celu przygotowania kontrolnego RNA, zmieszano 2 µ l Control RNA (Poly-A RNA Control Kit) z 38 µl wolnej od nukleaz wody do uzyskania całkowitej objętości 40 µ l.
Przygotowanie całkowitego RNA miało na celu uzyskanie dokładnej jego ilości (100 ng), w celu przeprowadzenia dalszej procedury. W tym celu, dokonano kilku rozcieńczeń według dołączonego przez firmę Affymetrix schematu.
Procedura przygotowania całkowitego RNA:
1. Dodano 2 µl Poly-A Control Stock do 38 µl buforu Poly-A Control Dil (pierwsze rozcieńczenie 1:20). Wymieszano i odwirowano.
2. Dodano 2 µ l pierwszego rozcieńczenia do 98 µ l buforu Poly-A Control Dil (drugie rozcieńczenie 1:50). Wymieszano i odwirowano.
3. Dodano 2 µl drugiego rozcieńczenia do 98 µ l buforu Poly-A Control Dil (trzecie rozcieńczenie 1:50). Wymieszano i odwirowano.
17
4. Dodano 2 µl trzeciego rozcieńczenia do 98 µl buforu Poly-A Control Dil (czwarterozcieńczenie 1:10). Wymieszano i odwirowano.
5. Dodano 2 µl czwartego rozcieńczenia do 100 ng całkowitego RNA.
3.2.2.b. Synteza pojedynczo-niciowego cDNA.
Procedura przygotowania materiału do analizy aktywności genów za pomocą mikromacierzy RNA składała się z kilku etapów, zgodnie z protokołem dołączonym przez producenta (Affymetrix), prowadzących do uzyskania pojedynczo-niciowego cDNA, który następnie poddano fragmentacji i znakowaniu.
3.2.2.c. Transkrypcja in vitro.
Z każdej próbki wcześniej przygotowanego całkowitego RNA pobrano po 1. µl materiału i uzupełniono 4. µl wody wolnej od nukleaz, a następnie zmieszano z odczynnikami znajdującymi się w zestawie First Strand Master Mix (The Ambion® WT Expression Kit) i inkubowano w termocyklerze UNO II (Biometra) w odpowiednich warunkach (tabela 1). Kolejno dodano po 50 µ l odczynników z zestawu Second-Strand Master Mix (The Ambion® WT Expression Kit) do każdej probówki i inkubowano w warunkach podanych w tabeli numer 1 - Cykl pierwszy, synteza drugiej nici cDNA.
Uzyskane w ten sposób dwu-niciowe cDNA poddano reakcji transkrypcji in vitro wykorzystując mieszaninę buforu oraz enzymów (IVT, The Ambion® WT Expression Kit) w warunkach opisanych w tabeli nr 2.
Tabela 1. Warunki inkubacji w termocyklerze podczas pierwszego etapu transkrypcji in vitro.
Program (metoda)
Temperatura pokrywy termocyklera
Etap 1 Etap 2 Etap 3
Cykl pierwszy, synteza
pierwszej nici cDNA 50 o C 25 o C, 60 min. 40 oC, 60 min. 4 oC, 2 min. Cykl pierwszy, synteza
drugiej nici cDNA -
16 oC, 60 min. 65 oC, 10 min. 4 oC, 2 min.
18
Tabela 2. Warunki inkubacji w termocyklerze podczas przygotowywania materiału do analizy
aktywności wybranych genów za pomocą mikromacierzy RNA.
Program (metoda) Temperatura pokrywy termocyklera Etap 1 Etap 2 Transkrypcja in vitro 50 o C 40 oC, 16 godz. 4 oC
3.2.2.d. Oczyszczanie cRNA oraz ocena ilościowo-jakościowa.
Otrzymany w wyniku transkrypcji in vitro jedno-niciowy RNA poddano procesowi oczyszczania, wykorzystując odpowiednie bufory z zestawu (The Ambion® WT Expression Kit). Zestaw ten zawierał złoża magnetyczne, które wiązały się z cRNA otrzymanym w procesie transkrypcji in vitro. W celu związania cRNA do złoża, dodano 60 µl uprzednio przygotowanej mieszaniny cRNA Binding Mix do każdej próbki i dobrze wymieszano. Umieszczono próbki na płytce o dołkach kształtu litery U. Dodano 60 µ l 100% izopropanolu do każdej próbki. Dobrze wymieszano. Delikatnie wytrząsano przez 2 minuty, a następnie przeniesiono płytkę z cRNA na podstawkę magnetyczną. Po około 5 minutach, płyn stał się przejrzysty, a cRNA utworzyło krążek w miejscu zetknięcia z magnesem. Delikatnie usunięto supernatant i przemyto cRNA za pomocą 100 µ l buforu myjącego (Nucleic Acid Wash Solution) z jednoczesnym wytrząsaniem przez 1 minutę, a następnie osuszono. Jeszcze raz powtórzono przemywanie. Dodano do każdej próbki 40 µl uprzednio ogrzanego buforu Elution Solution, inkubowano przez 2 minuty. Intensywnie wytrząsano płytkę przez 3 minuty, a następnie przeniesiono na magnetyczną podstawkę i usunięto supernatant, zawierający cRNA do nowej płytki wolnej od nukleaz. Zamrożono w temperaturze -20 oC.
Po zakończeniu oczyszczania cRNA, określono jego stężenie mierząc spektrofotometrycznie jego absorbcję przy długości fali 260 nm. Jakość uzyskanego cRNA sprawdzono na 1.5% żelu agarozowym z dodatkiem bromku etydyny.
3.2.2.e. Drugi cykl cDNA.
Kolejnym etapem procedury była synteza pojedynczo-niciowego cDNA na bazie uzyskanego wcześniej cRNA. W tym celu odpowiednią ilość cRNA (10 µg) zmieszano z mieszaniną starterów z zestawu (Random Primers, The Ambion® WT Expression Kit) i poddano denaturacji w warunkach: 5 minut w temperaturze 70 oC, 5 minut w temperaturze 25
o
19
Po denaturacji całość uzupełniono o mieszaninę reakcyjną zawierającą bufor oraz enzymy drugiego cyklu odwrotnej transkrypcji (2-nd-Cycle Master Mix), wymieszano energicznie, odwirowano i poddano inkubacji w termocyklerze w odpowiednich warunkach (tabela 3). Natychmiast po inkubacji próbki odwirowano.Tabela 3. Warunki inkubacji na etapie syntezy drugiego cyklu cDNA.
Program (metoda)
Temperatura pokrywy termocyklera
Etap 1 Etap 2 Etap 3 Etap 4
Cykl drugi, synteza cDNA 75 o C 25 o C, 10 min. 42 oC, 90 min. 70 oC, 10 min. 4 oC, 2 min. 3.2.2.f. Hydroliza, oczyszczanie oraz ocena ilościowo-jakościowa materiału.
Otrzymane w drugim cyklu cDNA poddano procesowi hydrolizy z wykorzystaniem RNazyH. Do każdej próbki dodano po 2 µl RNazyH, wymieszano, odwirowano, a następnie poddano inkubacji w warunkach przedstawionych w tabeli nr 4.
Tabela 4. Warunki hydrolizy cDNA.
Program (metoda)
Temperatura pokrywy termocyklera
Etap 1 Etap 2 Etap 3
Hydroliza 75 oC 37 oC, 45 min. 95oC, 5 min. 4 oC, 2 min.
Po zakończeniu hydrolizy przeprowadzono oczyszczanie otrzymanego pojedynczo-niciowego cDNA. W tym celu dodano 18 µ l wolnej od nukleaz wody do każdej próbki w celu uzyskania objętości końcowej 60 µ l. Następnie dodano 60 µl buforu cDNA Binding Mix do każdej próbki. Dobrze wymieszano. Przeniesiono próbki na płytkę o dołkach w kształcie litery U. Dodano 120 µl 100% etanolu do każdej próbki i dobrze wymieszano. Wytrząsano delikatnie przez 2 minuty. Umieszczono płytkę w magnetycznej podstawie. Po ok. 5. minutach, gdy płyn był klarowny, usunięto supernatant. Przemywano próbki dodając po 100 µ l mieszaniny myjącej (Nucleic Acid Wash Solution), wytrząsano przez 1. minutę, a następnie umieszczono płytkę na magnetycznej podstawce i usunięto supernatant. Przemywanie powtórzono. Umieszczono płytkę na wytrząsarce i wytrząsano przez 1. minutę w celu odparowania etanolu, po czym dodano do każdej próbki 30 µ l buforu Elution Solution, ogrzanego wcześniej do temperatury 55 – 58 oC. Inkubowano przez 2. minuty w temperaturze pokojowej. Intensywnie wytrząsano płytkę przez 3 minuty, umieszczono ją na magnetycznej
20
podstawie i po chwili zebrano supernatant do nowej, wolnej od nukleaz płytki. Po oczyszczeniu próbek określono ich stężenia na spektrofotometrze przy długości fali 260 nm oraz sprawdzono jakość na 1,5% żelu agarozowym z dodatkiem bromku etydyny.3.2.2.g. Fragmentacja cDNA.
W dalszym etapie przeprowadzono proces fragmentacji cDNA. Przygotowano bufor zawierający otrzymany wcześniej cDNA (cDNA Fragmentation Buffer) oraz bufor do fragmentacji (Fragmentation Master Mix). Do 31,2 µl buforu zawierającego cDNA dodano 16,8 µl buforu Master Mix. Delikatnie odwirowano, a następnie inkubowano w warunkach: 37 oC przez 60 minut, 93 oC przez 2 minuty, 4 oC przez minimum 2. minuty.
Odwirowano, a następnie przeniesiono 45 µl każdej badanej próbki do nowych probówek. Pofragmentowany materiał zamrożono w temperaturze – 20 oC.
3.2.2.h. Znakowanie cDNA.
Znakowanie pojedynczo-niciowego cDNA przeprowadzono w celu wizualizacji miejsca hybrydyzacji cDNA do odpowiednich sond na mikromacierzy. W tym celu przygotowano mieszaninę do znakowania (tabela 5) i wymieszano w odpowiednich proporcjach z pofragmentowanym cDNA (15 µl buforu do znakowania Labeling Master Mix i 45 µ l badanej próbki cDNA). Całość poddano inkubacji w warunkach: 37 oC przez 60 minut, 70 oC przez 10 minut, 4 oC przez 2 minuty.
Tak przygotowany materiał wykorzystano w procesie hybrydyzacji.
Tabela 5. Składniki buforu do znakowania Labeling Reaction Master Mix.
Składniki buforu
Labeling Master Mix Ilość na 1 macierz
Ilość na 4 macierze (z uwzględnieniem 10% nadwyżki) 5x bufor TdT 12 µl 52.8 µl TdT, 30 U/µl 2 µl 8.8 µl Odczynniki do znakowania DNA 1 µl 4.4 µl Objętość całkowita 15 µl 66 µl 3.2.2.i. Hybrydyzacja.
Pofragmentowane i znakowane cDNA wykorzystano w metodzie hybrydyzacji, zgodnie z procedurą dołączoną przez producenta, firmę Affymetrix (GeneAtlas TM WT Expression Kit User Manual, GeneAtlas TM Hybrydyzation, Wash, and Stain Kit for WT
21
Array Strip). Zgodnie z wytycznymi, przygotowano bufor do hybrydyzacji (WT Hybrydyzation Master Mix) zgodnie z poniższą tabelą.Tabela 6. Składniki buforu do hybrydyzacji.
Kolejność
podania Składniki buforu
Ilość na jedną macierz Ilość na 4 macierze (z 10% nadwyżką) 1 5X WT Hyb Add 1 30 µl 132 µl 2 Kontrolne oligonukleotydy B2 (3 nM) 1.5 µl 6.6 µl 3 20X kontrole hybrydyzacji
(bioB, bioC, bioD, cre) 7.5 µl 33 µl
4 15X WT Add 4 10 µl 44 µl
Całkowita objętość 49 µl 215.6 µl
Następnie dodano po 49 µl buforu do hybrydyzacji do każdego dołka w bloczku (4 macierze w jednym bloku), po czym dodano 41 µl pofragmentowanego i znakowanego (badanego) cDNA oraz 60 µl 2.5X buforu WT Hyb Add 6 do końcowej objętości 150 µ l. Wymieszano i odwirowano. Probówki z badanym materiałem poddano denaturacji w termocyklerze w temperaturze 99 oC przez 5 minut, a następnie w 45 oC przez 5 minut. W celu usunięcia z próbek jakichkolwiek składników nierozpuszczalnych, odwirowywano je w 5000 RPM przez 1. minutę w temperaturze pokojowej. Przygotowywano macierze do hybrydyzacji poprzez zanurzenie ich w 120 µ l buforu do hybrydyzacji. Istotnym było usunięcie wszelkich pęcherzyków powietrza. Przygotowane macierze umieszczono w stacji hybrydyzacyjnej (Hybridization Oven 645) w temperaturze 48 oC i rozpoczęto proces hybrydyzacji, który w przypadku materiału zwierzęcego (szczur) trwał 20 godzin. Warunki procesu hybrydyzacji (czas oraz temperaturę) kontrolowano za pomocą programu GeneAtlas™ Software Setup firmy Affymetrix. Po zakończeniu procesu hybrydyzacji przystąpiono do płukania bloków z macierzami.
3.2.2.j. Płukanie macierzy i odczyt wyników.
Kolejnym etapem procedury analizy ekspresji genów metodą mikromacierzy RNA było płukanie badanych macierzy w celu usunięcia buforu do hybrydyzacji oraz niezwiązanych cDNA. Cały proces przeprowadzono w stacji płukania Fluidics Station 450. Przygotowano bufory do płukania, ogrzano do temperatury pokojowej, delikatnie wymieszano. Podano odpowiednie bufory w określonych ilościach do odpowiednich dołków w płytce do
22
przemywania, według schematu umieszczonego w instrukcji obsługi GeneAtlas™ System User’s Guide. Umieszczono płytki do przemywania w stacji do przemywania i rozpoczęto procedurę. Czas oraz tempo przemywania kontrolowano za pomocą programu komputerowego GeneAtlas™ Instrument Control Software. Po zakończeniu płukania mikromacierze umieszczano w stacji odczytu (GeneAtlas™ Imaging Station) i rozpoczynano odczyt wyników za pomocą programu komputerowego GeneAtlas Viewer (Affymetrix), w wyniku czego utworzono wiele plików wynikowych z rozszerzeniem dat. Na każdą macierz została nałożona siatka w celu określenia dokładnego położenia wszystkich punktów, po czym przekonwertowano pliki dat do plików cel. Dodatkowo każdy wynik przedstawiono w postaci plików jpg (ryc. 2). Sumarycznie cała procedura wykonania doświadczenia mikromacierzowego została przedstawiona na rycinie 1.Ryc. 1. Sumaryczna procedura wykonywania doświadczenia z zastosowaniem mikromacierzy firmy
23
Ryc. 2. Zdjęcie mikromacierzy RNA po odczytaniu wyników za pomocą programu GeneAtlas
Viewer.
3.2.2.k. Analiza danych uzyskanych metodą mikromacierzy RNA.
Analizę mikromacierzy RNA przeprowadzono z wykorzystaniem kilku programów komputerowych. W pierwszym etapie zastosowano dostarczony przez producenta program Partek Express Affymetrix Edition. Za jego pomocą przeprowadzono kontrolę jakości hybrydyzacji i znakowania oraz kontrolę jakości użytego RNA. Próby pogrupowano (po 3 powtórzenia na grupę), a następnie porównano je do kontroli. W programie wykorzystano test parametryczny ANOVA (test rozkładu normalnego). Po analizie otrzymane wartości p (p-value) zostały poddane korekcie za pomocą FDR (False Discovery Rate). Dla każdego genu otrzymano wykresy, w których na osi Y przedstawiono poziom ekspresji w skali logarytmicznej (Log2 intensity), na osi X poszczególne dni regeneracji. Wyniki umieszczono w tabeli, w której dla każdego genu podano opis, symbol, numer dostępu Genebanku oraz wartości p i zmianę ekspresji względem kontroli (fold change, cut-off). Do dalszych analiz wykorzystano pakiet Bioconductor, będący składową języka R. W tym celu ze strony internetowej Bioconductor pobrano odpowiednie biblioteki do analizy danych oraz bibliotekę opisową dla macierzy szczura (pd.ragene.1.1.st.v1), w której znajduje się opis lokalizacji poszczególnych genów. Wczytano pliki cel, a następnie przeprowadzono korektę tła oraz normalizację za pomocą algorytmu RMA (ang. Robust Multiarray Analysis). Po ukończonym procesie normalizacji, uzyskane wyniki scalono z plikiem opisowym, w którym znajdują się nazwy oraz charakterystyki funkcji genów. Dla każdej grupy genów obliczono średni poziom ekspresji, a następnie jej zmianę względem kontroli. Przeprowadzono testy statystyczne
24
ANOVA z uwzględnieniem FDR, co zostało wyeksportowane do tabeli (plik csv). Dane z pliku csv wykorzystano do wyselekcjonowania genów różniących się istotnie statystycznie (p<0.05), których ekspresja różniła się względem kontroli przynajmniej dwukrotnie (fold>+-2) przynajmniej w jednej badanej grupie (w jednym badanym dniu regeneracji). Wyselekcjonowane geny przedstawiono na wykresie słupkowym, z zaznaczeniem genów, których ekspresja wzrastała lub malała względem kontroli (p<0.05, zmiana ekspresji >2 lub <2). Wykorzystując bazę danych Gene Ontology (GO), wszystkie wyselekcjonowane geny podzielono na grupy: geny związane z angiogenezą, geny związane z proliferacją oraz geny związane ze steroidoegnezą. Poszczególne grupy genów przedstawiono na wykresach punktowych (scatter plot) oraz wykresach słupkowych. Następnie geny te podzielono na klastry odzwierciedlające podobny profil ekspresji przy zastosowaniu algorytmu klasteryzacji hierarchicznej. Klastry przedstawiono w postaci wykresów „map cieplnych” (heat maps).Analiza funkcjonalna uzyskanych wyników została przeprowadzona z zastosowaniem pakietu języka R – GeneAnswer. Pakiet ten między innymi pozwala na zbadanie przynależności wyselekcjonowanych wcześniej genów do poszczególnych procesów biologicznych. Uzyskane geny pogrupowano poprzez kategorie funkcjonalne bazy ontologii genów – Gene Ontology – Biological Proces (GO.BP). Pakiet GeneAnswer umożliwia również zbadanie istotności zmian ekspresji genów należących do danej grupy ontologicznej, poprzez strategię wzbogacania grupy genów (gene set enrichment) oraz wykorzystanie testu hipergeometrycznego. Wszystkie kategorie funkcjonalne z p<0.05 testu hipergeometrycznego zaznaczono zielonym tłem.
Wyniki uzyskane metodą mikromacierzy RNA poddano walidacji metodą reakcji łańcuchowej polimerazy w czasie rzeczywistym (real time PCR, QPCR).
25
3.2.3. Walidacja wyników otrzymanych metodą mikromacierzy RNA za pomocą
metody QPCR.
3.2.3.a. Odwrotna transkrypcja.
Wyizolowany uprzednio całkowity RNA poddano odwrotnej transkrypcji z zastosowaniem polimerazy AMV firmy QIAGEN. Przygotowano mieszaninę składającą się z: 1 µg RNA, 1
µl startera (dT)18, wody wolnej od nukleaz (do końcowej objętości 10 µl).
Mieszaninę inkubowano przez 5 minut w temperaturze 70 oC, a następnie schładzano na lodzie. W zależności od liczby badanych prób przygotowywano mieszaninę reakcyjną w odpowiedniej proporcji. Dla pojedynczej próby jej skład był następujący: 4 µl buforu reakcyjnego, 1 µl 20 U/µl inhibitora RNaz, 2 µl 10 mM dNTP, 2 µl 200 U/µl odwrotnej transkryptazy AMV.
Tę samą procedurę, z zastąpieniem odwrotnej transkryptazy taką samą objętością wody wolnej od nukleaz, przeprowadzano dla prób kontrolnych (-RT). Reakcje przeprowadzano w temperaturze 4 oC w czasie 60 minut z użyciem termocyklera UNO II (Biometra).
3.2.3.b. QPCR.
Reakcję PCR w czasie rzeczywistym (QPCR lub Real-Time PCR) przeprowadzono na urządzeniu LightCycler 2.0 firmy ROCHE. W przeprowadzeniu tej metody wykorzystano barwnik SYBR Green jako detektor, który oddziałując z dwu-niciowym DNA emituje fluorescencję. Dokonano również analizy krzywej topnienia otrzymanych produktów.
Startery zaprojektowano z wykorzystaniem programu Primer 3 (tabela 7). Program reakcji QPCR obejmował kilka etapów. Pierwszym etapem była 10-minutowa denaturacja, a następnie przeprowadzano właściwą reakcję PCR, która składała się z 45. cykli: denaturacja w temperaturze 95 oC przez 10 sekund, przyłączanie startera w temperaturze 60 oC przez 5 sekund, wydłużanie w temperaturze 72 oC przez 7 sekund.
26
Tabela 7. Sekwencje starterów wykorzystane w reakcji RT-PCR oraz w celu walidacji wyników
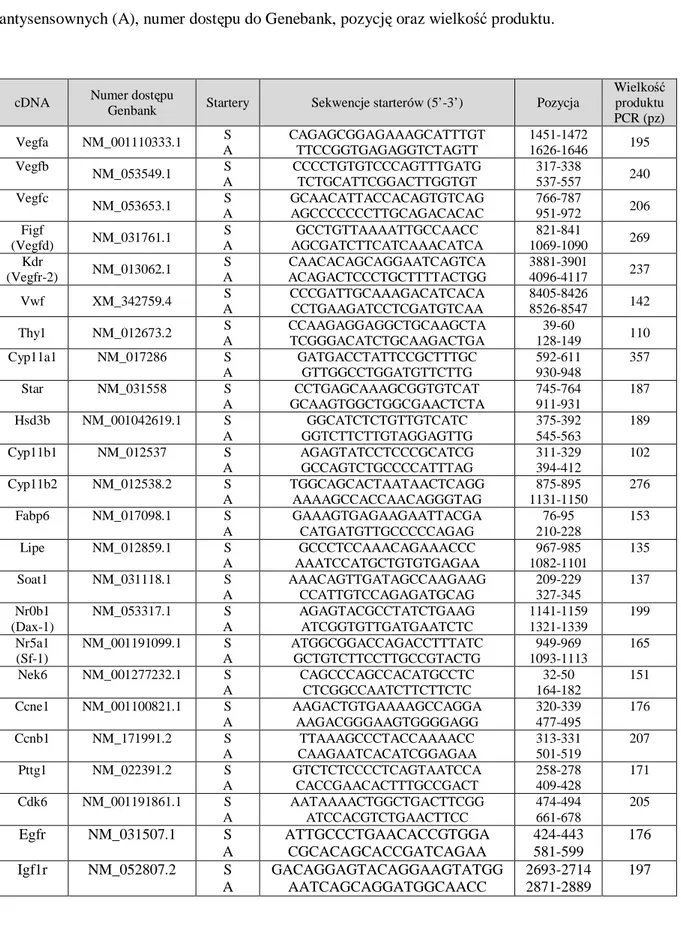
mikromacierzy RNA metodą QPCR. W tabeli przedstawiono sekwencje starterów sensownych (S), antysensownych (A), numer dostępu do Genebank, pozycję oraz wielkość produktu.
cDNA Numer dostępu
Genbank Startery Sekwencje starterów (5’-3’) Pozycja
Wielkość produktu PCR (pz) Vegfa NM_001110333.1 S A CAGAGCGGAGAAAGCATTTGT TTCCGGTGAGAGGTCTAGTT 1451-1472 1626-1646 195 Vegfb NM_053549.1 S A CCCCTGTGTCCCAGTTTGATG TCTGCATTCGGACTTGGTGT 317-338 537-557 240 Vegfc NM_053653.1 S A GCAACATTACCACAGTGTCAG AGCCCCCCCTTGCAGACACAC 766-787 951-972 206 Figf (Vegfd) NM_031761.1 S A GCCTGTTAAAATTGCCAACC AGCGATCTTCATCAAACATCA 821-841 1069-1090 269 Kdr (Vegfr-2) NM_013062.1 S A CAACACAGCAGGAATCAGTCA ACAGACTCCCTGCTTTTACTGG 3881-3901 4096-4117 237 Vwf XM_342759.4 S A CCCGATTGCAAAGACATCACA CCTGAAGATCCTCGATGTCAA 8405-8426 8526-8547 142 Thy1 NM_012673.2 S A CCAAGAGGAGGCTGCAAGCTA TCGGGACATCTGCAAGACTGA 39-60 128-149 110 Cyp11a1 NM_017286 S A GATGACCTATTCCGCTTTGC GTTGGCCTGGATGTTCTTG 592-611 930-948 357 Star NM_031558 S A CCTGAGCAAAGCGGTGTCAT GCAAGTGGCTGGCGAACTCTA 745-764 911-931 187 Hsd3b NM_001042619.1 S A GGCATCTCTGTTGTCATC GGTCTTCTTGTAGGAGTTG 375-392 545-563 189 Cyp11b1 NM_012537 S A AGAGTATCCTCCCGCATCG GCCAGTCTGCCCCATTTAG 311-329 394-412 102 Cyp11b2 NM_012538.2 S A TGGCAGCACTAATAACTCAGG AAAAGCCACCAACAGGGTAG 875-895 1131-1150 276 Fabp6 NM_017098.1 S A GAAAGTGAGAAGAATTACGA CATGATGTTGCCCCCAGAG 76-95 210-228 153 Lipe NM_012859.1 S A GCCCTCCAAACAGAAACCC AAATCCATGCTGTGTGAGAA 967-985 1082-1101 135 Soat1 NM_031118.1 S A AAACAGTTGATAGCCAAGAAG CCATTGTCCAGAGATGCAG 209-229 327-345 137 Nr0b1 (Dax-1) NM_053317.1 S A AGAGTACGCCTATCTGAAG ATCGGTGTTGATGAATCTC 1141-1159 1321-1339 199 Nr5a1 (Sf-1) NM_001191099.1 S A ATGGCGGACCAGACCTTTATC GCTGTCTTCCTTGCCGTACTG 949-969 1093-1113 165 Nek6 NM_001277232.1 S A CAGCCCAGCCACATGCCTC CTCGGCCAATCTTCTTCTC 32-50 164-182 151 Ccne1 NM_001100821.1 S A AAGACTGTGAAAAGCCAGGA AAGACGGGAAGTGGGGAGG 320-339 477-495 176 Ccnb1 NM_171991.2 S A TTAAAGCCCTACCAAAACC CAAGAATCACATCGGAGAA 313-331 501-519 207 Pttg1 NM_022391.2 S A GTCTCTCCCCTCAGTAATCCA CACCGAACACTTTGCCGACT 258-278 409-428 171 Cdk6 NM_001191861.1 S A AATAAAACTGGCTGACTTCGG ATCCACGTCTGAACTTCC 474-494 661-678 205 Egfr NM_031507.1 S A ATTGCCCTGAACACCGTGGA CGCACAGCACCGATCAGAA 424-443 581-599 176 Igf1r NM_052807.2 S A GACAGGAGTACAGGAAGTATGG AATCAGCAGGATGGCAACC 2693-2714 2871-2889 197
27
cDNA Numer dostępu
Genbank Startery Sekwencje starterów (5’-3’) Pozycja
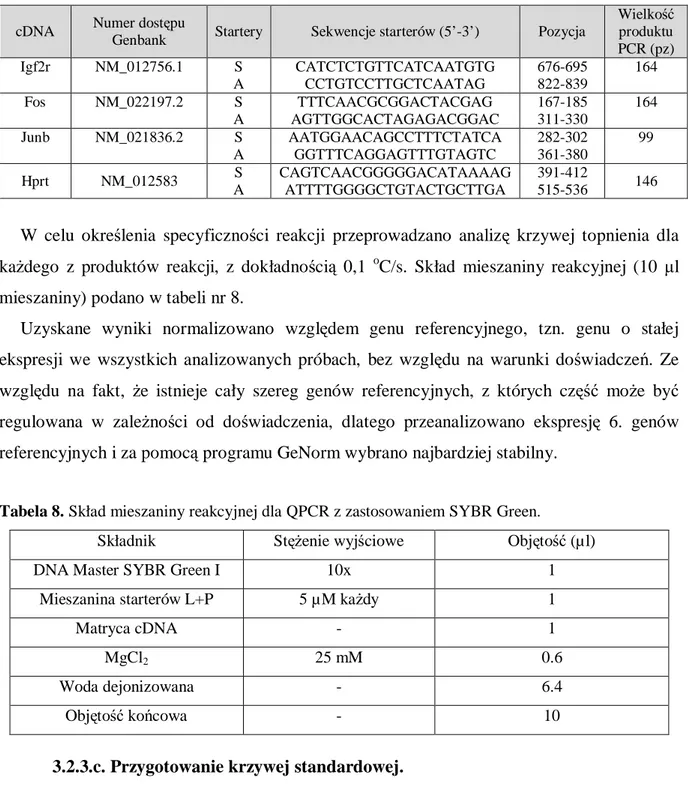
Wielkość produktu PCR (pz) Igf2r NM_012756.1 S A CATCTCTGTTCATCAATGTG CCTGTCCTTGCTCAATAG 676-695 822-839 164 Fos NM_022197.2 S A TTTCAACGCGGACTACGAG AGTTGGCACTAGAGACGGAC 167-185 311-330 164 Junb NM_021836.2 S A AATGGAACAGCCTTTCTATCA GGTTTCAGGAGTTTGTAGTC 282-302 361-380 99 Hprt NM_012583 S A CAGTCAACGGGGGACATAAAAG ATTTTGGGGCTGTACTGCTTGA 391-412 515-536 146
W celu określenia specyficzności reakcji przeprowadzano analizę krzywej topnienia dla każdego z produktów reakcji, z dokładnością 0,1 oC/s. Skład mieszaniny reakcyjnej (10 µl mieszaniny) podano w tabeli nr 8.
Uzyskane wyniki normalizowano względem genu referencyjnego, tzn. genu o stałej ekspresji we wszystkich analizowanych próbach, bez względu na warunki doświadczeń. Ze względu na fakt, że istnieje cały szereg genów referencyjnych, z których część może być regulowana w zależności od doświadczenia, dlatego przeanalizowano ekspresję 6. genów referencyjnych i za pomocą programu GeNorm wybrano najbardziej stabilny.
Tabela 8. Skład mieszaniny reakcyjnej dla QPCR z zastosowaniem SYBR Green.
Składnik Stężenie wyjściowe Objętość (µl)
DNA Master SYBR Green I 10x 1
Mieszanina starterów L+P 5 µM każdy 1
Matryca cDNA - 1
MgCl2 25 mM 0.6
Woda dejonizowana - 6.4
Objętość końcowa - 10
3.2.3.c. Przygotowanie krzywej standardowej.
W celu określenia efektywności reakcji PCR w czasie rzeczywistym dla każdego z badanych genów przygotowano krzywą standardową. Dlatego, wykorzystano produkty klasycznej reakcji RT-PCR, które rozdzielono na 2% żelu agarozowym, a następnie wyizolowano z żelu przy użyciu DNA Gel Extraction Kit (MILLIPORE). Przy użyciu spektrofotometru określano ilość uzyskanego w taki sposób DNA. Poprzez powielenie seryjnych dziesięciokrotnych rozcieńczeń uzyskanego DNA utworzono krzywe standardowe. Warunkach tych reakcji były dokładnie takie same, jakie ustalono dla badanych próby.
28
3.2.4. Badanie immunohistochemiczne.
3.2.4.a. Dwustopniowa metoda immunoperoksydazowa z kompleksem EnVision-HRP.
W celu lokalizacji peptydów w skrawkach regenerujących nadnerczy wykorzystano dwustopniową reakcję immunohistochemiczną wzmocnioną polimerowyn kompleksem sprzężonym z peroksydazą chrzanową (EnVision-HRP, DAKO). Materiał, który stanowiły regenerujące nadnercza, pobierano zaraz po dekapitacji odpowiednio po 1., 2., 3., 5., 8. i 15. dniu od enukleacji. Tkanki utrwalano w płynie Bouina przez 24h, następnie odwadniano i zatapiano w parafinie. Grubość skrawków wynosiła 5 – 6 µm. Następnie preparaty odparafinowywano i doprowadzano do fazy wodnej. W zależności od specyfikacji przeciwciał, skrawki poddawano lub nie poddawano gotowaniu. Etap gotowania przeprowadzano w buforze cytrynianowym (pH 9.0 lub pH 6.0), umieszczając skrawki w sumie na 9 minut w mikrofalówce (2 razy na 3.5 minuty oraz 1 raz na 2 minuty). Następnie preparaty schładzano do temperatury pokojowej. Dalsze etapy przeprowadzano w wilgotnej komorze w temperaturze pokojowej. W celu zahamowania aktywności endogennej peroksydazy skrawki inkubowano w 3% roztworze perhydrolu przez 10 minut. Kolejno preparaty preinkubowano w roztworze surowicy koziej (w stosunku 1:20 w PBS z 0,1% BSA i 0,1% azydkiem sodu) w temperaturze pokojowej przez 30 minut w celu zablokowania miejsc niespecyficznie wiążących przeciwciało i tym samym zmniejszając ryzyko wystąpienia tła. Po tak przeprowadzonej inkubacji odsączano surowicę kozią za pomocą bibuły, a następnie nakładano swoiste przeciwciało skierowane przeciw odpowiednim antygenom. Na kontrolne skrawki nakładano rozpuszczalnik przeciwciała (PBS z 0,1% BSA i 0,1% azydkiem sodu). Preparaty inkubowano w odpowiednich warunkach, w zależności od użytego przeciwciała (przez noc w temperaturze 4 oC lub w temperaturze pokojowej przez 2 godziny). Po inkubacji preparaty dokładnie przepłukiwano w roztworze PBS 3 razy po 3 minuty z wytrząsaniem (wytrząsarka PST-60 HL plus, Thermo Shaker) i inkubowano przez 30’ w obecności drugorzędowego przeciwciała sprzężonego z polimerem znakowanym peroksydazą chrzanową. Następnie preparaty płukano w PBS (3 razy po 3 min) i nakładano na nie roztwór 3, 3’-diaminobenzydyny (DAB) – substratu peroksydazy (DAKO). Po około 40. - 60. sekundach skrawki przepłukiwano w wodzie destylowanej, część z nich podbarwiano hematoksyliną. Następnie odwadniano je przeprowadzając przez szereg alkoholi o rosnącym stężeniu (alkohol etylowy 70%, 80% i 90%, alkohol absolutny –2 razy, alkohol z ksylenem w stosunku objętościowym 1:1, ksylen – 3 razy), zatapiano w syntetycznym olejku do montowania preparatów (Permount, Fisher Scientific) i nakładano szkiełka nakrywkowe.
29
3.2.4.b. Analiza jakościowa preparatów immunohistochemicznych.
Uzyskane preparaty immunohistochemiczne analizowano z wykorzystaniem mikroskopu optycznego (BX40 Olympus). Preparaty zeskanowano za pomocą skanera Pannoramic MIDI firmy 3DHISTECH Ltd., a zdjęcia wykonano i przeanalizowano za pomocą programu Mirax Viewer (Zeiss).
3.2.5. Analiza statystyczna wyników.
Wyniki przedstawiono w postaci średnich arytmetycznych ± SE (błąd standardowy średniej). Analizę statystyczną przeprowadzono przy użyciu analizy wariancji (ANOVA), a następnie wielokrotnego testu rozstępu Duncana lub testu t-Studenta. W obliczeniach posługiwano się programem Statistica.
30
4. Wyniki
4.1. Profil globalnej ekspresji genów w toku indukowanej enukleacją regeneracji
nadnerczy szczura.
Metodą mikromacierzy ekspresyjnych przeanalizowano ekspresję ponad 29. tysięcy genów w toku indukowanej enukleacją regeneracji nadnercza szczura, w 1., 2., 3., 5., 8. i 15. dniu po zabiegu enukleacji. Taką samą analizę przeprowadzono w nadnerczach kontrolnych. Wyniki ekspresji genów porównywano do nadnerczy kontrolnych przy założeniu zmiany ekspresji >2 (wzrost/spadek) oraz wartości p<0,05. Odnotowano, iż w pierwszym dniu po enukleacji 1966 genów wykazało zmianę ekspresji, przy czym aż 1256 genów charakteryzował wzrost ekspresji, podczas gdy spadek ekspresji zanotowano w przypadku 710. genów (ryc. 3). Najwyższy wzrost ekspresji w tym czasie wykazywały geny związane ze stanem zapalnym, odpowiedzią na stres, odpowiedzią immunologiczną, adhezją, migracją komórek, powstawaniem i organizacją skrzepu (ryc. x). Na przykład, do tej grupy należały geny kodujące metalopeptydazy macierzy pozakomórkowej, fibronektynę, receptory chemokin i ich ligandy, interleukiny, czy też integryny. Na końcu badanego okresu regeneracji (w 15. dniu doświadczenia) wysoką ekspresję odnotowano w przypadku genów związanych z procesami metabolicznymi w komórkach regenerującej kory nadnercza (np., Fabp6, białko wiążące kwasy tłuszczowe i inne ligandy hydrofobowe, które może uczestniczyć w metabolizmie steroidów w komórkach produkujących hormony steroidowe).
W okresie między 1. a 3. dniem regeneracji najwyższą ekspresję odnotowano w przypadku genów związanych z rozwojem zapalenia i tworzenia skrzepu. Na przykład, ekspresja genu Serpina3n (NM_031531), który koduje glikoproteinę działającą jak inhibitor peptydazy serynowej w 1. dniu po enukleacji wzrosła 84 razy w porównaniu do nadnerczy kontrolnych. Podobnie, odnotowano 47-krotny wzrost ekspresji genu Vcan (NM_001170558, który koduje białko macierzy zewnątrzkomórkowej, uwalniane przez wiele typów komórek). Ponadto, ekspresja genów Prg4 (NM_001105962), Chi3l1 (NM_053560) i Fcgr2b (NM_175756) była około 40 razy wyższa w porównaniu do nadnerczy kontrolnych. Z kolei, odnotowano spadek ekspresji Mc2r (receptora ACTH) w 1. dniu regeneracji, który nie uległ zmianie w późniejszym okresie doświadczenia.
31
Ryc. 3. Liczba genów wykazujących w ciągu 15. dni zmianę ekspresji (wzrost/spadek) w odniesieniu
do grupy kontrolnej, w przebiegu indukowanej enukleacją regeneracji nadnerczy szczura przy założeniu: zmiany ekspresji >2 i p<0,05. Kolor niebieski – wzrost ekspresji, biały – spadek ekspresji.
Przeprowadzono również analizę mikromacierzy RNA mającą na celu pogrupowanie genów biorących udział w określonych procesach biologicznych (analiza klasteryzacji). Grupowano tylko geny - na podstawie danych bazy GO, których ekspresja w toku indukowanej enukleacją regeneracji kory nadnerczy szczura ulegała zmianie (wzrost/spadek >2, p<0,05; w stosunku do grupy kontrolnej). W każdej kategorii przyprowadzony został hipergeometryczny test statystyczny, a wyniki przedstawiono na ryc. 4. Stwierdzono, że liczba tych genów stopniowo spadała w toku regeneracji nadnercza szczura. W pierwszym dniu po enukleacji, ze wszystkich 1591 genów, 254 geny związane były z odpowiedzią na zranienie, 467 genów – z odpowiedzią na stres, 215 – z odpowiedzią obronną, 176 – z migracją komórek, podczas gdy 170 genów związanych było z adhezją komórek, a tylko 70 z podziałami mitotycznymi. 48 godzin po enukleacji, najbardziej znaczące statystycznie zmiany ekspresji dotyczyły genów związanych z odpowiedzią na zranienie (216 genów), migracją komórek (153 genów), mobilnością komórek (159 genów), gojeniem ran (125 genów), procesami układu odpornościowego (266 genów) i odpowiedzią na stres (392 genów). W trzecim dniu regeneracji największe różnice w ekspresji zanotowano w przypadku genów związanych z cyklem komórkowym (155 genów) oraz podziałem mitotycznym. W okresie między 5. a 15. dniem regeneracji nadal najbardziej znaczące zmiany ekspresji odnotowano w przypadku genów związanych z odpowiedzią na zranienie, stres, odpowiedzią immunologiczną, migracją leukocytów, a także adhezją komórek.
32
Ryc. 4. Analiza mikromacierzy RNA w oparciu o analizę ontologiczną (GO). Geny, których ekspresja
ulegała zmianom (wzrost/spadek >2, p<0,05; w odniesieniu do grupy kontrolnej) w toku regeneracji kory nadnerczy szczura pogrupowano na podstawie udziału w określonych procesach biologicznych na kategorie funkcyjne. Analizę oparto o dane zawarte w bazie danych GO, wykorzystując pakiet GeneAnswer programu R. Po lewej stronie umieszczono kategorie funkcyjne, na dole – ogólną liczbę genów, których ekspresja ulegała zmianie w danym dniu doświadczenia, u góry – dni regeneracji. Kolor zielony oznacza istotne statystcznie zmiany ekspresji (p<0,05). Im wartość p jest niższa, tym kolor zielony jest ciemniejszy.
33
4.2. Angiogeneza w indukowanej enukleacją regeneracji nadnercza szczura.
4.2.1. Analiza ekspresji genów związanych z angiogenezą.
Geny związane z angiogenezą wybrane zostały z wykorzystaniem bazy danych GO. Słowem kluczowym przy selekcji genów był wyraz „angiogeneza”. Wynik analizy ekspresji genów związanych z angiogenezą w trakcie indukowanej enukleacją regeneracji nadnercza szczura metodą mikromacierzy RNA przedstawiono za pomocą wykresu słupkowego oraz punktowego (ang. scater plot), na których przedstawiono liczbę genów, których ekspresja uległa zmianie, przy założeniu: zmiany ekspresji >2 oraz p<0,05, w stosunku do grupy kontrolnej (ryc. 5 i 6).
W 1. dniu regeneracji kory nadnerczy szczura wzrost ekspresji odnotowano w przypadku 40. genów, kodujących między innymi: fibronektynę 1 (Fn1, NM_019143), zasadowy czynnik wzrostu fibroblastów (Fgf2, NM_019305), interleukinę 1b (Il1b, NM_031512), czynnik wzrostu śródbłonka typu D (Figf, NM_031761.1), a spadek ekspresji – 12. Natomiast, w 15. dniu regeneracji nadnercza zmiany ekspresji odnotowano już tylko w przypadku 10. genów (6 wykazało wzrost ekspresji i 4 – spadek).
Ryc. 5. Liczba genów związanych z angiogenezą w toku indukowanej enukleacją regeneracji
nadnerczy szczura wykazujących zmiany ekspresji (wzrost/spadek) przy założeniu zmiany ekspresji >2 i p<0,05, w stosunku do grupy kontrolnej. Kolor niebieski – wzrost ekspresji, biały – spadek ekspresji.
34
Ryc. 6. Wykresy punktowe ekspresji genów związanych z angiogenezą w toku indukowanej
enukleacją regeneracji nadnerczy szczura w porównaniu z kontrolą przy założeniu: zmiana ekspresji >2 i p<0,05. Linie pomarańczowe oznaczają zmianę ekspresji, górna lewa część wykresu oznacza wzrost ekspresji, dolna prawa – spadek ekspresji.
Wyniki analizy mikromacierzy przedstawiono także za pomocą mapy „cieplnej“, na której geny związane z angiogenezą, przy założeniu p<0,05 i zmianie ekspresji >2 (w stosunku do grupy kontrolnej), poddano analizie klasteryzacji hierarchicznej, uzyskując tym samym 5 grup genów o wspólnym profilu ekspresji (ryc. 7). Klasteryzacji dokonano na tzw. „surowych danych” wartości ekspresji uzyskanej z plików CEL mikromacierzy. Wartość ekspresji przeskalowano do wartości skali Z (Z-score; zakres od -3 do +3), wysoką ekspresję genu oznaczono kolorem zielonym, przy czym maksymalna wartość oznaczono jako +3 (intensywny kolor zielony). Niską wartość ekspresji przedstawiono kolorem czerwonym, przy czym minimalna wartość oznaczono jako -3 (intensywny kolor czerwony). Kolorem czarnym oznaczono geny których wartość ekspresji mieściła się w środku pomiędzy wartością maksymalną a minimalną. Kolor czarny nie oznacza, że ekspresja danego genu równa jest kontroli. W każdej grupie znajdują się zarówno geny kodujące czynniki pobudzające, jak i hamujące angiogenezę. Uzyskano następujące grupy genów: 1 - geny, których najwyższa ekspresja przypadła między 8. a 15. dniem doświadczenia (tylko 4 geny); 2 - geny, w przypadku których najwyższy poziom ekspresji odnotowano między 3. a 5. dniem regeneracji; 3 – geny z najwyższą ekspresją w grupie kontrolnej; 4 - geny, w przypadku których zanotowano wysoką ekspresję w pierwszych dniach regeneracji; 5 - geny, których najwyższy poziom ekspresji odnotowano w pierwszym dniu po enukleacji. Poziom ekspresji niektórych genów przedstawiono w postaci wykresów kulkowych (ryc. 8), gdzie dynamiczne
35
zmiany ekspresji są lepiej widoczne. Na ryc. 8. przedstawiono profil ekspresji Vegfc (przedstawiciel grupy 3.), Figf (grupa 2.) i Thy1 (grupa 2). Ponadto, na wykresie przedstawiono także profil ekspresji genów, które nie zostały wyselekcjonowane w bazie GO, a są znane ze swego udziały w angiogenezie: CD34 – marker śródbłonka i receptor Plaur (NM_134352), aktywator plazminogenu, receptor urokinazy. Dodatkowo, pokazano także profil ekspresji genów o znanej proangiogennej funkcji (Vegfa i Vwf), których ekspresja nie uległa zmianie w toku regeneracji nadnercza. Spośród nowo zidentyfikowanych czynników, przeanalizowano ekspresję angiopoetyn (Ang). Poziom mRNA angiopoetyny 1 (Ang-1) obniżył się w trakcie doświadczenia, podczas gdy poziom ekspresji angiopoetyny 2 (Ang-2) wzrósł w 2. i 5. dniu regeneracji. Przeciwnie, ekspresja genu receptora angiopoetyn (Tek, Tie2) pozostała niezmieniona w toku regeneracji.36
Ryc. 7. Analiza ekspresji genów związanych z angiogenezą w toku indukowanej enukleacją
regeneracji nadnerczy szczura metodą mikromacierzy RNA przedstawiona za pomocą mapy „cieplnej”. Wykres przedstawia grupy genów o wspólnym profilu ekspresji, przy założeniu: zmiany ekspresji >2 i p<0,05, w stosunku do grupy kontrolnej. Klasteryzacji dokonano na tzw. „surowych danych” wartości ekspresji uzyskanej z plików CEL mikromacierzy. Wartość ekspresji przeskalowano do wartości skali Z (Z-score; zakres od -3 do +3), wysoką ekspresję genu oznaczono kolorem zielonym, przy czym maksymalna wartość oznaczono jako +3 (intensywny kolor zielony). Niską wartość ekspresji przedstawiono kolorem czerwonym, przy czym minimalna watość oznaczono jako -3 (intensywny kolor czerwony).
37
Ryc. 8. Profil ekspresji wybranych genów związanych z angiogenezą w toku indukowanej enukleacją
regeneracji nadnerczy szczura przedstawiony w postaci wykresów kulkowych, przy założeniu zmiany ekspresji >2 i p<0,05, w stosunku do grupy kontrolnej (analizowane dane pochodzą z „mapy cieplnej”. Poszczególne geny reprezentują 5 grup genów przedstawionych na mapie cieplnej – 1 (Fgf9), 2 (Thy1), 3 (Vegfc), 4 (Fgf2), 5 (Il1b).
Spośród wszystkich genów związanych z angiogenezą w toku indukowanej enukleacją regeneracji nadnercza szczura, których profil ekspresji przeanalizowano metodą mikromacierzy RNA, wybrano kilka genów o najwyższej ze wszystkich ekspresji. Należą do nich geny kodujące czynniki bezpośrednio i pośrednio uczestniczące w formowaniu nowo powstających naczyń krwionośnych, takie jak: czynnik wzrostu Vegfa, czynnik wzrostu Figf, receptor 2 Vegf (Kdr, NM_013062.1), a także czynnik Vwf (XM_342759.4) oraz antygen powierzchniowy Thy1. Profil ekspresji tych genów określony metodą QPCR omówiono w dalszej części rozprawy.
38
4.2.2. Walidacja metodą QPCR ekspresji wybranych genów.
Wyniki ekspresji wybranych genów uzyskane metodą mikromacierzy RNA zostały poddane walidacji metodą QPCR.
Spośród genów związanych z angiogenezą, w celu walidacji wyników metodą QPCR wybrano kilka, których ekspresja po analizie mikromacierzy ekspresyjnych była najwyższa, przy założeniu zmiany ekspresji >2 oraz p<0,05, w stosunku do grupy kontrolnej. Ponadto, ze względu na fakt, iż do analizy ekspresji metodą mikromacierzy, w bazie GO nie zostały wybrane ważne geny o dobrze znanej proangiogennej funkcji, dodatkowo przebadano też metodą QPCR ekspresję takich genów. Wśród genów związanych z angiogenezą w toku indukowanej enukleacją regeneracji nadnerczy szczura, których ekspresję analizowano, znalazły się: czynnik wzrostu komórek śródbłonka typu A, typu D, receptor 2 Vegf, czynnik Von Willebranda oraz antygen powierzchniowy komórek Thy1.
Na ryc. 9. przedstawiono profil ekspresji badanych genów. Wykazano, iż poziom mRNA genu Vegfa i Kdr w pierwszym dniu regeneracji był bardzo niski w porównaniu z kontrolą, w następnych natomiast dniach eksperymentu nie różnił się od kontroli. Ponadto, najwyższy poziom ekspresji genu Figf zanotowano w 5. dniu doświadczenia, podczas gdy genu Vwf w pierwszym i drugim dniu. Ekspresja genu Figf w ciągu całego badanego okresu regeneracji utrzymywała się na znacząco wyższym poziomie w stosunku do kontroli. Między 2. a 5. dniem regeneracji nadnerczy szczura wzrósł znacząco poziom transkryptu Thy1, po czym spadł, utrzymując się jednak stale na wyższym poziomie w porównaniu z kontrolą. Ekspresja genu Srpx2 (produkty białkowe którego biorą udział w migracji komórek śródbłonka), znacząco zwiększa się w toku regeneracji.
39
Ryc. 9. Półilościowa ocena ekspresji wybranych genów (Vegfa, Figf, Kdr, Srpx2, Vwf i Thy1)
związanych z angiogenezą w toku indukowanej enukleacją regeneracji nadnerczy szczura. Metoda PCR w czasie rzeczywistym. Słupki przedstawiają względną ekspresję badanych genów w stosunku do ekspresji genu Hprt, średnia z trzech oznaczeń, z zaznaczonym SE. Różni się od grupy kontrolnej: * - p < 0,05; ** - p < 0,02; *** - p < 0,01; **** - p < 0,001.